ERJ:肺动脉高压的遗传咨询和检测—国际PAH遗传研究联盟共识
2022-11-23 肺动脉高压研究进展 肺动脉高压研究进展

虽然PAH是一种主要具有常染色体显性遗传模式的疾病,但也有少数患者在不同的基因中存在一个以上的致病突变,并可能对疾病的表现有所贡献。
虽然PAH是一种主要具有常染色体显性遗传模式的疾病,但也有少数患者在不同的基因中存在一个以上的致病突变,并可能对疾病的表现有所贡献。例如,在一个家族中,有一个BMPR2移码突变和一个EIF2AK4外显子跳跃突变,这些所谓的寡基因病例就被描述出来。早期的一项研究报道了一个患有BMPR2和KCNA5突变体的儿童的严重PAH病例。因此,在少数情况下,两个突变体可能会增加疾病的严重性或导致PAH发病年龄提前。然而,这种潜在的寡源性遗传对PAH的贡献仍有待确定。
在PAH患者中,对现有治疗反应的巨大差异是否至少部分由遗传倾向决定,目前并不清楚。十多年前就有研究表明,BMPR2变异体携带者很少对血管扩张剂有急性反应,因此不太可能从钙通道阻滞剂治疗中获益。相对来说,很少有研究关注可能与PAH疗法的药物反应相互作用的基因突变。
最近有两个病例系列探讨了BMPR2基因的致病变体可能与吸入或肠外前列腺素的血流动力学反应降低有关的假设。结论是不同的,但值得注意的是,不同的结果可能与治疗方法的积极性有关。这些努力最终可能为精准医学在PAH中的应用提供了一个步骤。在这方面,确定具有遗传风险的患者是否对新出现的药物表现出不同的反应将是有趣的,这些药物旨在通过BMP途径(如sotatercept)或血小板衍生生长因子受体途径(如吸入式酪氨酸激酶抑制剂seralutinib)减少细胞增生和血管重塑。
新生儿持续性肺动脉高压患者常伴有复杂的合并症或疾病,并可能怀有发育基因的遗传突变。除了公认的PAH基因如TBX4和BMPR2外,这些基因对患者表型的确切影响还有待确定。先天性心脏病PAH患者可能表现为不同的心脏表型。特定的心脏缺陷对PAH的贡献有多大,或者某些基因对心脏和肺血管发育是否有多效作用,仍有待确定。在先天性心脏病患者中,那些早期修复的患者更有可能存在致病突变。
PAH遗传咨询推荐
至少应向被诊断为IPAH、FPAH、PVOD/PCH和厌食症诱发的和先天性心脏病相关的疑似患者提供遗传咨询和基因检测。
应采用PAH基因组测序方法,至少包括所有强有力的证据PAH基因ACVLR1、ATP13A3、BMPR2、CAV1、EIF2AK4、ENG、GDF2、KCNK3、KDR、SMAD9、SOX17、TBX4。随着更多的基因被发现和以前描述的可能的疾病基因有了新的支持性证据,该基因列表应该被修订。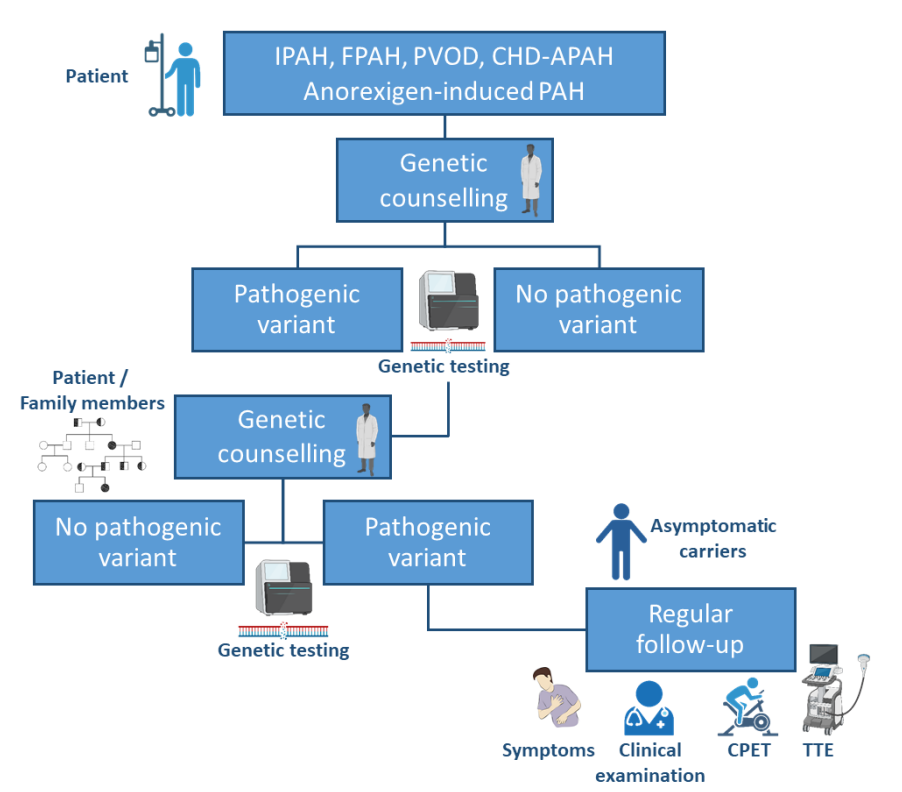 图1,PAH患者及其亲属的遗传咨询路径。在进行基因检测前后,患者应接受遗传咨询。如果确定了一个(可能的)致病突变,应鼓励进行家族测试。如果发现无症状的携带者,应定期进行临床随访。
图1,PAH患者及其亲属的遗传咨询路径。在进行基因检测前后,患者应接受遗传咨询。如果确定了一个(可能的)致病突变,应鼓励进行家族测试。如果发现无症状的携带者,应定期进行临床随访。
测试方法应辅以量化读数的方法,以评估基因内或整个基因的缺失或复制,特别是BMPR2、ACVRL1、ENG和TBX4等基因。
有症状的FPAH患者、PAH遗传正常的儿童和先天性异常的患者,无论年龄大小,都应考虑进行外显子/基因组测序,包括FPAH的多个受影响的家庭成员和儿童的父母双方,以提高识别致病突变的机会。
在确定有PAH相关致病/可能致病突变的家庭中,应与遗传专业人员协商提供级联基因检测。对无症状的遗传高危人群应定期随访,并了解PAH的相关症状,以便早期诊断PAH,并在出现PAH的证据时尽早开始治疗。应定期对意义不确定的变体进行重新评估,以考虑到新发表的数据。PH专家可以监测ClinVar以了解特定变体的进一步信息,或联系实验室要求重新评估。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



