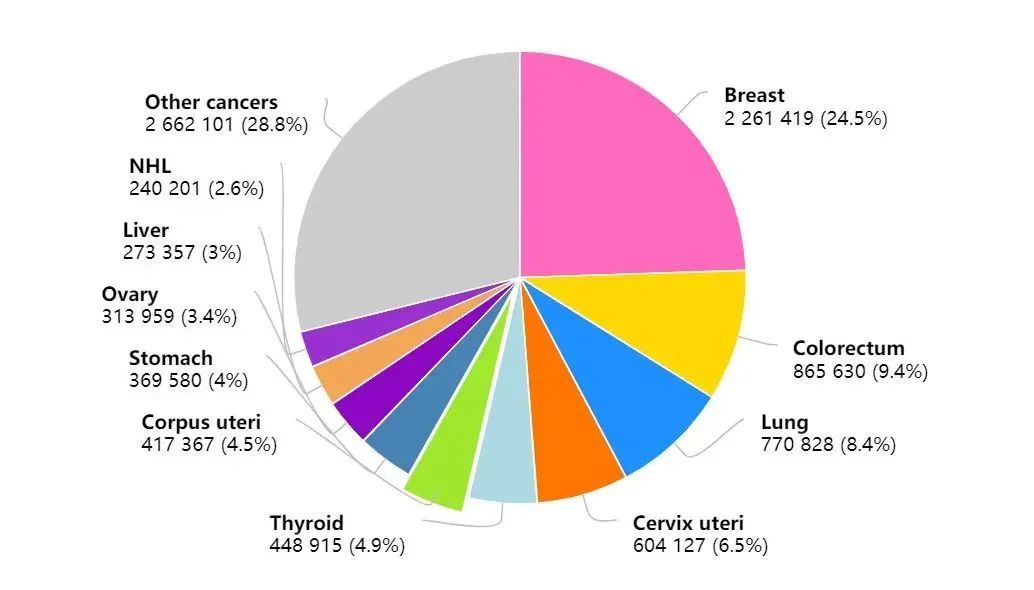
综述:选择性CDK抑制剂的研究进展
2022-11-10 精准药物 精准药物

CDK是一类丝氨酸/苏氨酸激酶,在细胞周期调节及细胞转录过程中发挥关键作用。CDK活性的失调与肿瘤的发生发展密切相关,以CDK为靶点设计抗肿瘤药物是一大研究热点。
CDK是一类丝氨酸/苏氨酸激酶,在细胞周期调节及细胞转录过程中发挥关键作用。CDK活性的失调与肿瘤的发生发展密切相关,以CDK为靶点设计抗肿瘤药物是一大研究热点。0在上一篇文章中,我们总结了泛CDK抑制剂的临床研究进展。泛CDK抑制剂由于治疗窗狭窄,许多相关的临床试验已被终止。因此,选择性CDK抑制剂的开发至关重要,本文总结了选择性CDK抑制剂的设计策略、构效关系和治疗应用。
CDK1抑制剂
CDK1选择性抑制剂可以可逆地阻断细胞周期的G2/M期,并在早期有丝分裂中实现细胞同步化。
 图:Avotaciclib的设计策略、化学结构和CDK抑制活性
图:Avotaciclib的设计策略、化学结构和CDK抑制活性
Avotaciclib(BEY1107, 18)是一种可口服的CDK1抑制剂,目前正处于I/II期临床试验中,作为单一治疗药物或与吉西他滨联合用药,用于治疗局部晚期或转移性胰腺癌(NCT03579836)。这种化合物不仅可以控制癌细胞的细胞分裂,还可以通过消除癌症干细胞的靶向机制来克服抗癌药物的耐受性。当与细胞毒性药物吉西他滨联合使用时,avotaciclib有望克服吉西他滨新出现的耐药性。CDK2抑制剂
CDK2/细胞周期蛋白E复合物在视网膜母细胞瘤(Rb)蛋白磷酸化和G1/S期过渡中起重要作用。CDK2/细胞周期蛋白A复合物在S期的DNA合成和G2/M期过渡中CDK1/细胞周期蛋白B的活化中起重要作用。PF-07104091(19)是一种口服可利用的CDK2抑制剂,具有潜在的抗肿瘤活性。在人类卵巢癌细胞模型(OVCAR3, OV5392)中,化合物19(25, 75和175mg/kg, 一天两次, po)可以以剂量依赖性的方式诱导肿瘤减小。此外,19和一线药物帕博西尼的联合给药在乳腺癌细胞模型中表现出协同作用。辉瑞公司于2020年开始一项化合物19的I/II期临床试验,以评估化合物19的安全性和耐受性,并在小细胞肺癌,非小细胞肺癌,卵巢癌或乳腺癌患者中测试化合物19作为单一治疗药物的最大耐受剂量(NCT04553133)。
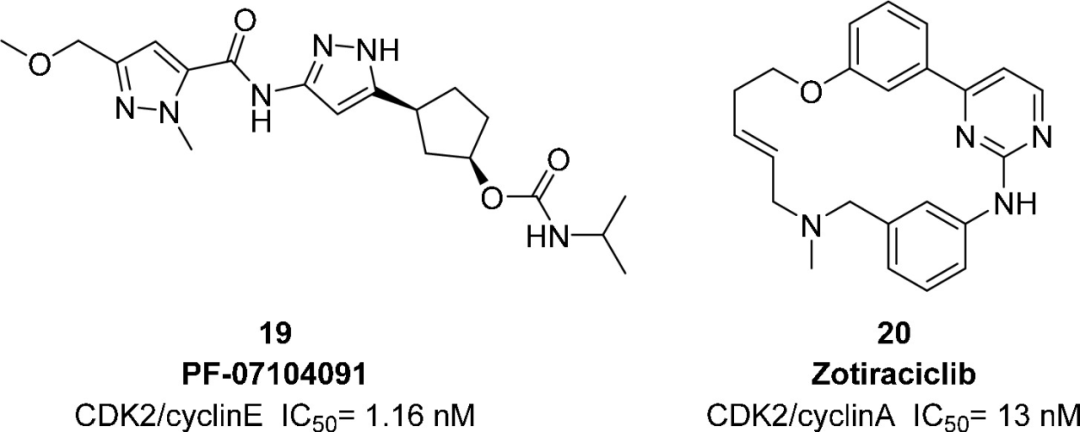 图:PF-07104091(19)和zotiraciclib(20)的结构和CDK抑制活性
图:PF-07104091(19)和zotiraciclib(20)的结构和CDK抑制活性
Zotiraciclib(TG02,20)是一种独特的具有大环结构的分子。大环的构象有利于在与蛋白质结合时呈现出关键的药效团。此外,具有碱性氮的大环与CDK2活性位点入口处的酸性残基Asp86形成盐桥,这也解释了化合物20的高活性。作为一种高效口服的CDK2抑制剂,化合物20可以穿透血脑屏障,降解MCL-1和Myc癌蛋白。Myc蛋白的过表达是人HGG和弥漫性内生型桥脑胶质瘤(DIPG)的特征之一。2021年1月,完成了化合物20的Ib期临床试验(HHSN261200800001E)。此外,化合物20和替莫唑胺联合用于治疗复发性HGG患者显示出良好的耐受性和疗效。FDA和欧洲药品管理局(EMA)已批准化合物20作为孤儿药用于治疗胶质瘤。化合物20目前正在进行两项独立的I期临床试验,用于治疗多形性胶质母细胞瘤(GBM)和DIPG(NCT03904628, NCT03224104)。
CDK4/6抑制剂
近年来,乳腺癌的发病率和死亡率持续上升。通过确定肿瘤的雌激素受体(ER),黄体酮受体(PR)和HER2状态,乳腺癌可分为不同亚型。CDK4和CDK6是HR+/HER2-型乳腺癌发生和发展的重要驱动因素。CDK4/6是包括ER在内的多种促进生长的信号通路的常见下游靶标。在ER+乳腺癌中,雌激素信号传导导致ER-细胞周期蛋白D1-CDK4/6通路的活性增加,驱动细胞从G1期进入S期,最终导致不受控的细胞增殖。CDK4/6抑制剂可抑制CDK4/6与细胞周期蛋白D之间复合物的形成,阻断ATP结合,从而切断上游生长信号,阻止G1期向S期的过渡。目前由FDA批准的CDK抑制剂都是CDK4/6抑制剂:辉瑞的帕博西尼(PD-0332991, 21),诺华的瑞博西尼(LEE011, 22),礼来的阿贝西利(LY-2835219,23)和G1 Therapeutics的曲拉西利(G1T28,24)。CDK4和CDK6的ATP结合结构域表现出很高的结构相似性,这些结构域之间的差异仅在于Glu144(CDK4)和Gln149(CDK6)。这四种抑制剂都是ATP竞争性抑制剂,可以与激酶的ATP结合域结合。

吡啶并[2, 3-d]嘧啶-7-酮衍生物25是一种CDK4/6抑制剂,优化其结构得到化合物26(PD0183812)。该化合物具有良好的CDK4/6活性,但其对CDK亚型的选择性未达到预期。对母核C-2处氨基取代的研究显示,2-氨基吡啶取代化合物的选择性优于苯胺基取代物。随后,将母核替换为喹唑啉,得到化合物27a,27b,吡啶环被苯环取代,并在C-4处引入甲基,得到高选择性化合物27c-f,但新化合物的活性仍然不高。在母核的C-3处引入乙酰基团得到27g-i,这些化合物表现出非常好的活性和选择性。进一步优化,在C-2侧链上引入哌嗪、高哌嗪、4-羟基哌啶和吗啉等不同含氮烷烃,得到化合物21,对耐药CDK4/6表现出抑制活性。21在多种肿瘤类型中表现出临床活性,包括套细胞淋巴瘤,乳腺癌和脂肪肉瘤。21与他莫昔芬联合用药对ER+/HER2-乳腺癌的治疗具有协同作用。此外,21与芳香酶抑制剂(阿那曲唑和来曲唑, NCT02942355, NCT05190094)或选择性ER降解剂(氟维司群, NCT02738866, NCT04318223)的联合用药已在II/III期临床试验中进行了评估,表现出一定的临床优势。 图:帕博西尼(21)和SHR-6390(28)的设计策略、化学结构、结构修饰和CDK抑制活性
图:帕博西尼(21)和SHR-6390(28)的设计策略、化学结构、结构修饰和CDK抑制活性
用哌啶环取代哌嗪环得到了SHR-6390(28)。与21相比,口服化合物28消除了ER+和HER2+型乳腺癌对内分泌治疗的耐药性和HER2-靶向抗体的耐药性。此外,28与内分泌治疗相结合,对ER+乳腺癌表现出协同抗肿瘤活性。21和CDK6的共晶结构表明,嘧啶环与铰链区的氨基酸相互作用,并与Val101形成两个氢键。环戊基和疏水袋中的多种氨基酸残基形成疏水相互作用。4位甲基占据Phe80形成的狭长疏水腔。哌嗪环延伸到溶剂区域,并在ATP结合位点的溶剂暴露区域与氨基酸残基Asp102和lle19形成极性相互作用。CDK6的晶体结构表明,左侧的氨基嘧啶形成两个氢键,酰胺羰基形成一个氢键。环戊基在疏水口袋中形成疏水相互作用。哌嗪延伸到溶剂区形成盐桥。虽然23的结构发生了重大变化,但23与CDK6的共晶结构表明其关键结合特性与之前批准的CDK4/6抑制剂基本相同。氨基嘧啶仍然形成两个氢键,并且吡啶仍然存在。蛋白与21和22中的羰基形成的氢键丧失,但与咪唑形成新的氢键。化合物23中的异丙基可以模仿化合物21和22中的环戊基,产生疏水作用。对22的酰胺取代基进行环化,与吡咯并[2, 3-d]嘧啶母核形成三环,环戊烷取代基变为环己烷,得到化合物24,推测其结合模式与22相似。 图:帕博西尼(21),瑞博西尼(22),阿贝西利(23)和CDK6的相互作用
图:帕博西尼(21),瑞博西尼(22),阿贝西利(23)和CDK6的相互作用
化合物22和23被批准为HR+/HER2-转移性乳腺癌的一线治疗药物,它们与非甾体芳香酶抑制剂(阿那曲唑和来曲唑),他莫昔芬或戈舍瑞林等药物的联合用药正处于研究阶段。此外,它们作为单一疗法以及与化疗药物联合用药用于治疗其他肿瘤(包括黑色素瘤、晚期实体瘤、淋巴瘤和神经母细胞瘤)的临床研究也在进一步开展。化合物24是FDA批准的第一种“突破性疗法”,旨在改善接受化疗的癌症患者的预后。24可以在化疗前预防性地暂时阻断骨髓细胞的G1期,这将显著减少化疗药物对骨髓细胞的杀伤,从而保护骨髓细胞和免疫系统。
贝达药业在化合物23的苯并咪唑母核中并入一个饱和环烷烃,得到候选化合物BPI-16350(29),正在进行I期临床试验(NCT03791112),用于治疗晚期实体瘤患者。此外,研究人员开发了GLR-2007(30),用于治疗对标准临床疗法反应不佳的晚期复发性或难治性实体瘤,包括非小细胞肺癌(NSCLC)和GBM。目前正在进行I/II期临床试验,用于治疗晚期实体瘤(NCT04444427)。该药物已被批准作为孤儿药用于恶性胶质瘤的治疗,并且通过了治疗胶质母细胞瘤的快速通道审核。对30进行进一步修饰,得到lerociclib(G1T38, 31)。化合物31与氟维司群联合用药的研究处于I/II期临床实验,用于治疗内分泌衰竭后HR+/HER2-局部进展性或转移性乳腺癌患者(NCT02983071)。ON-123300(32)是CDK4/6和AMPK相关蛋白激酶5(ARK5)的双重抑制剂,用于晚期癌症的潜在治疗。化合物32的I期临床试验已经启动,用于治疗复发性癌症或对至少一种先前治疗耐药(包括对批准的第二代CDK4/6抑制剂耐药的转移性HR+/HER2-乳腺癌)的晚期癌症患者(NCT04739293)。FLX-925(33)是一种新型的双重抑制剂,靶向FMS样酪氨酸激酶-3(FLT-3)和CDK4/6,处于早期临床研究中,用于治疗复发性或难治性AML。一项评估化合物33治疗复发或难治性AML患者的安全性、药代动力学、药效学的I期临床研究正在进一步开展中(NCT02335814)。
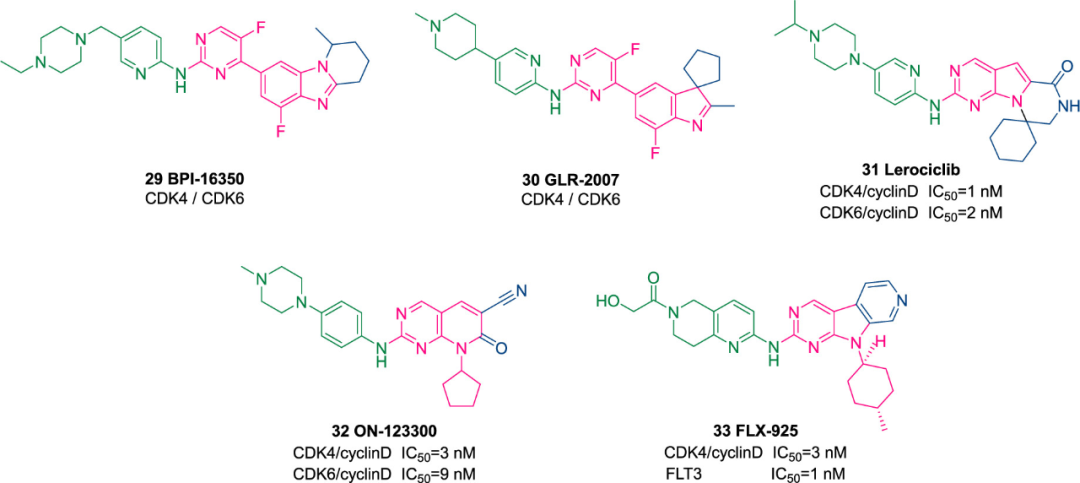 图:化合物29-33的结构和CDK抑制活性
图:化合物29-33的结构和CDK抑制活性
此外,正处于临床试验中的化合物34-41的结构尚未公开。
 CDK7抑制剂
CDK7抑制剂
CDK7同时参与转录和细胞周期的调节,在许多类型的癌症中过表达,还与侵袭性临床病理特征和预后不良相关。已进入临床试验的几种选择性CDK7抑制剂在许多临床前模型中显示出良好的抗癌效果,包括AML,前列腺癌,肝癌,胃癌,宫颈癌,胰腺癌,结直肠癌,神经母细胞瘤,NSCLC和SOX-2扩增的肺鳞状细胞癌。
吡唑并嘧啶衍生物samuraciclib(ICEC0942,42)可逆地结合CDK7的ATP结合位点(IC50=40nM),并在一系列癌症中表现出抗增殖活性,包括AML,小细胞肺癌(SCLC)和TNBC。吡唑并[1, 5-a]嘧啶母核与铰链区Leu83的主链之间存在两个氢键相互作用,哌啶环的氮原子与Asn132之间形成第三个氢键,前面的片段也与Asp145之间存在静电接触。化合物42良好的药代动力学特性使得该化合物成为有希望的临床候选药物。与他莫昔芬联合用药时,42完全阻断ER+异种移植瘤的生长,目前正在进行I/II期临床试验,用于治疗乳腺癌和前列腺癌(NCT03363893)。
 图:选择性CDK7抑制剂42-45的结构和CDK抑制活性
图:选择性CDK7抑制剂42-45的结构和CDK抑制活性
由于在ATP结合口袋外存在半胱氨酸和赖氨酸残基,也开发了许多共价CDK7抑制剂。共价结合的CDK抑制剂通常活性和选择性更高,治疗效果更长久,严重的副作用也更少。THZ1(43)含有N-苯基嘧啶-2-胺母核,丙烯酰胺与附近的Cys312不可逆结合,芳香部分嵌入CDK7内部,这表明43的吲哚环旋转,使其指向而不是位于开放的核苷酸结合口袋深处。化合物43可以在体外和体内抑制许多类型的肿瘤,如骨肉瘤、食管鳞状细胞癌、成人T细胞白血病、MYCN扩增的神经母细胞瘤、黑色素瘤和SCLC,但43在体内的半衰期短(在小鼠血浆中为45分钟),限制了临床开发。
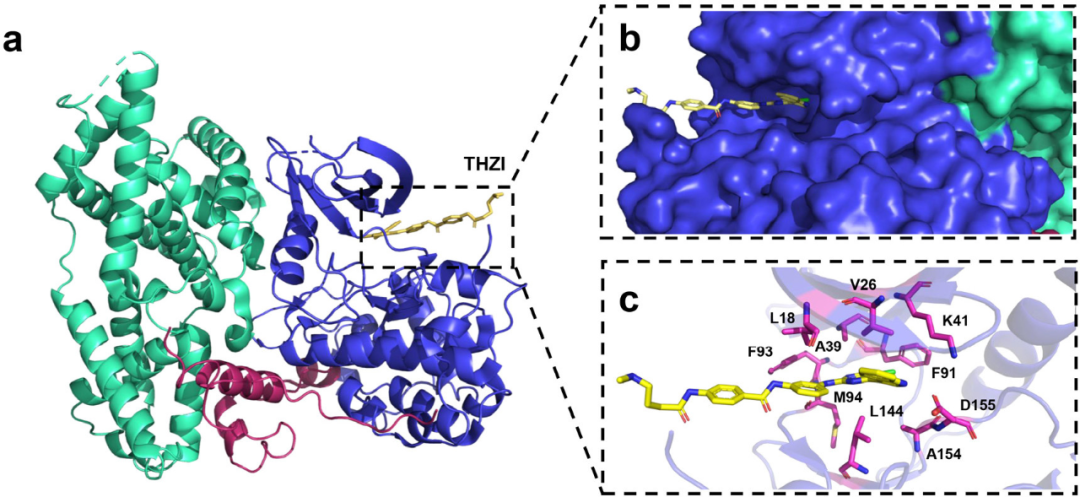 图:CAK-THZ1的结合模式(CAK:由CDK7、细胞周期蛋白H和MAT1组成的复合物)
图:CAK-THZ1的结合模式(CAK:由CDK7、细胞周期蛋白H和MAT1组成的复合物)
由化合物43衍生的CDK7抑制剂SY-1365(44),活性,选择性和代谢稳定性均有所改善。44的1, 3-环己烷二胺母核改善了形状互补性和疏水性,因此表现出比43更高的活性和选择性。化合物44通过增加酰胺连接子周围的空间位阻来限制弹头的构象,已进入I期临床试验,用于治疗卵巢癌和乳腺癌(NCT03134638)。然而,Syros医药最近宣布停止44的临床开发,优先推进一种新型选择性的非共价CDK7抑制剂SY-5609(45)。除氨基嘧啶铰链结合区外,(S)-3氨基哌啶环的碱性氮可与CDK7的ASP97形成盐桥,CDK7的ASP97是CDK家族中是保守的。用三氟甲基取代可提高活性,但略微降低选择性。因此,在吲哚环的C-6处引入氰基以向P环突出,得到的化合物选择性更高,这可能是利用了与其他CDK的P环的差异导致的。此外,为了改善药代动力学性质,由于其强氢键和高稳定性,引入了氧化膦部分。在化合物中引入偕二甲基可增加亲脂性,调节理化性质和渗透性,并提高口服生物利用度和选择性。结合模式表明哌啶6位可以为非保守的Val100提供更好的载体,因此C-6位引入偕二甲基,得到化合物45。化合物45与氟维司群的联合用药在卵巢癌,TNBC和ER+乳腺癌的临床前模型中显示出抗肿瘤活性。2020年初启动了一项化合物45治疗晚期实体瘤患者的I期试验(NCT04247126)。
此外,XL-102(AUR-102, 46, 结构未公开)是Exelixis早期临床开发的口服共价CDK7抑制剂,用于治疗实体瘤,正在进行一项I期临床研究(NCT04726332)。
CDK8/19抑制剂
CDK8及其旁系同源CDK19(97%同源性)可调节RNA PolI I的活性,在一些癌症中观察到CDK8/CDK19过表达。最新研究表明,CDK8和CDK19也可能在细胞重编程中发挥作用。CDK8/CDK19的靶向抑制可以激活自然杀伤(NK)细胞的抗癌作用。SEL120-34A(47)是一种新型可口服的苯并咪唑三环类的CDK8/19抑制剂,在AML的原位异种移植模型中,化合物47抑制肿瘤生长,缓解脾肿大并促进部分骨髓恢复。2020年,该化合物被FDA授予孤儿药资格,用于治疗AML。此外,正在开展一项评估47在转移性或晚期实体瘤患者中的安全性、药代动力学和疗效的I/II期研究(NCT05052255),47用于治疗AML或高风险MDS患者的试验正处在Ib期阶段(NCT04021368)。BCD-115(48)也是一种CDK8/19抑制剂,已进入治疗ER+/HER2-局部进展性和转移性乳腺癌的I期临床试验(NCT03065010)。
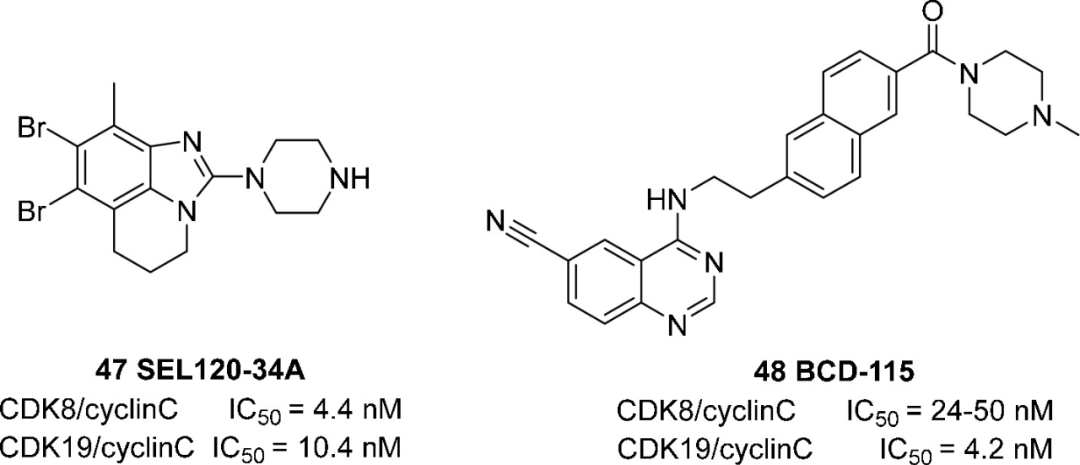 图:选择性CDK8/19抑制剂的结构和CDK抑制活性
图:选择性CDK8/19抑制剂的结构和CDK抑制活性
CDK9抑制剂
研究人员通过基于黄酮碱类泛-CDK抑制剂的修饰开发了一些选择性CDK9抑制剂,旨在提高选择性,降低胃肠道毒性和改善口服生物利用度。TP-1287(49)是化合物1的一种口服生物可利用的磷酸盐前药,可有效抑制CDK9。化合物49靶向并结合CDK9,这种结合降低了CDK9靶基因的表达,例如抗凋亡基因MCL-1,并在G1期诱导细胞周期停滞和CDK9过表达癌细胞中的细胞凋亡。一项多中心的I期剂量递增研究表明,49可导致磷酸化RNA PolI I水平的剂量依赖性降低,这与CDK9抑制的结果一致。化合物49给药后,在一名肉瘤患者中观察到局部响应,一名肾细胞癌(RCC)患者和两名膀胱癌患者的病情趋于稳定,基于这些可观的结果,49可作为单一疗法进行进一步的临床开发(NCT03604783)。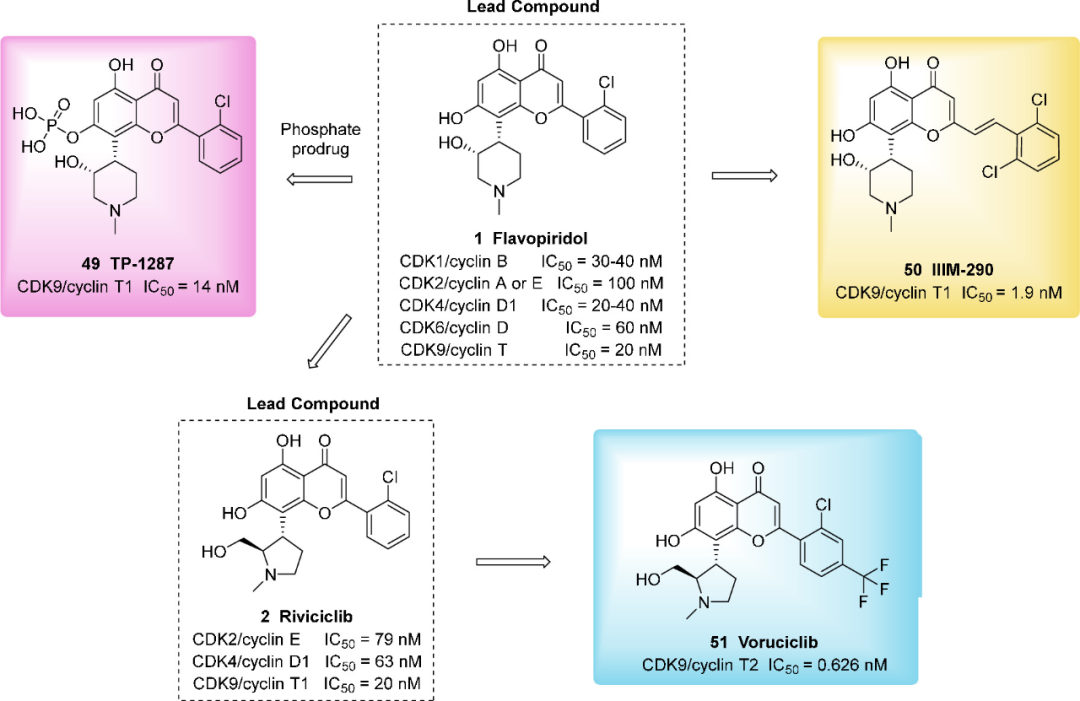 图:选择性CDK9抑制剂49-51的设计策略
图:选择性CDK9抑制剂49-51的设计策略
IIIM-290(50)是另一种新型可口服的选择性CDK9抑制剂。黄酮碱类化合物SAR的研究发现,在母核C-2位进行修饰,可以实现更高的选择性性和活性。化合物50是通过2, 6-二氯苯乙烯基取代C-2上的2-氯苯基得到的,这提高了其对CDK9的选择性,抑制活性也增加10倍。
C-2位苯乙烯环上卤素取代基的类型和位置与CDK9的选择性和抑制活性密切相关。氯取代比氟取代显示出更有效的活性。与对位取代相比,氯的邻位取代活性更高。结合模式表明,50占据了CDK9的ATP结合腔,色原酮母核,羟基哌啶和氯取代的苯乙烯是与CDK9的ATP结合位点结合的关键片段。化合物50在多个异种移植模型中显示出优异的药代动力学性质、口服生物利用度和有效的抗癌活性,在胰腺癌模型活性最好。目前已提交了50的IND申请,正在进行一项治疗局部进展性或转移性胰腺癌患者的I/II期临床试验(TENDER no.:12(318)/2021-P)。
将三氟甲基引入化合物2苯环的对位,得到voruciclib(P1446A-05, 51)。51作为单一疗法或与KRAS抑制剂(索托拉西布和阿达格拉西布)联合用药已在KRAS突变的癌细胞系显示出初步活性。2014年,暂停了51治疗复发或难治性CLL患者的临床研究(NCT02117336)。目前51正处于另一项I期临床试验中,以评估其在复发或难治性B细胞恶性肿瘤和标准疗法治疗后的AML患者中的安全性和初步疗效(NCT03547115)。
在药物设计中,砜基亚胺可以丰富结构多样性,提高代谢稳定性,优化理化性质以及引入氢键受体/供体。具有砜基亚胺基团的Atuveciclib(BAY-1143572,52)是第一个进入临床开发的口服选择性CDK9抑制剂。化合物52在小鼠和大鼠的多个异种移植瘤模型显示出单药治疗的良好疗效。已经完成了两项I期临床试验,评估52在晚期恶性肿瘤(NCT01938638)和晚期急性白血病(NCT02345382)患者中的安全性,耐受性,药代动力学和最大耐受剂量。将三嗪替换为吡啶,得到BAY-1251152(53)。与52相比,53表现出更好的活性(生化IC50=3nM, 细胞IC50=29nM),一项针对晚期癌症患者的I期剂量递增研究表明,53表现出可控的安全性、靶标药效学活性和抗肿瘤活性(NCT02635672)。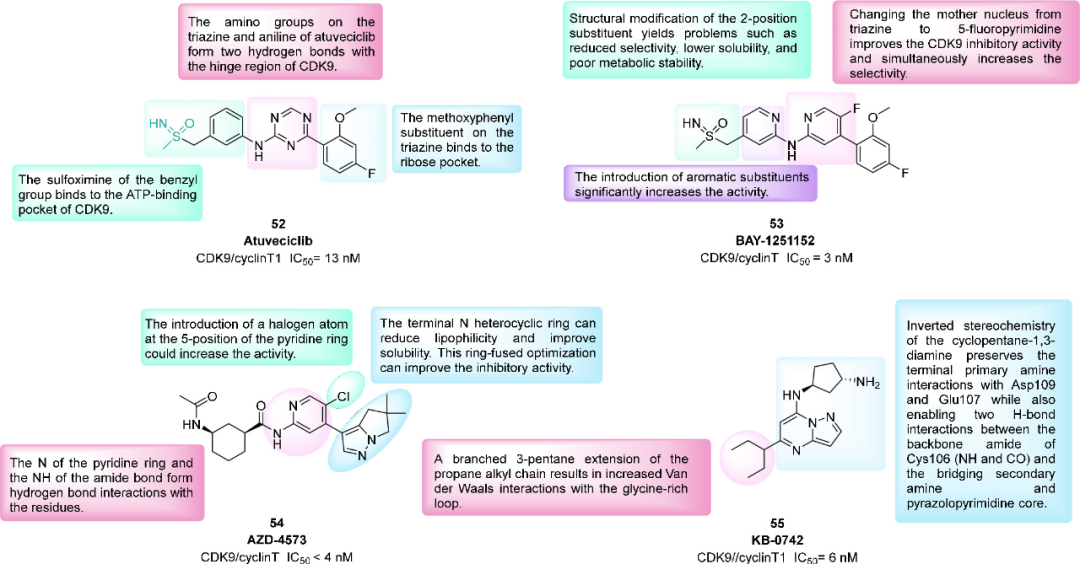 图:选择性CDK9抑制剂52-55的结构和活性
图:选择性CDK9抑制剂52-55的结构和活性
AZD4573(54)是一种高选择性(选择性>10倍)的CDK9抑制剂,可抑制MCL-1并诱导血液癌细胞凋亡。研究发现,末端苯环被杂环取代时,物理和化学性质得到改善,但活性丧失。将卤素引入吡啶环以补偿失去的活性,所得化合物54在活性和理化性质之间实现了总体平衡。54可导致MM,AML和非霍奇金淋巴瘤(NHL)异种移植瘤模型小鼠的肿瘤持续消退。正在进行一项治疗晚期血癌患者的I/II期临床试验(NCT04630756)。
KB-0742(55)是一种可口服的选择性CDK9抑制剂,对CDK9(IC50=6nM)的选择性超过50倍。在去势抵抗性前列腺癌(CRPC)细胞模型中,55可以通过抑制CDK9活性来降低癌基因Myc的表达。该化合物具有较长的血浆半衰期,并表现出剂量依赖性的靶标抑制活性,在体内模型也表现出良好药效。正在进行一项I期临床试验,用于治疗复发或难治性实体瘤或NHL患者(NCT04718675)。
GFH009(56, 结构未公开)是有效的CDK9抑制剂,选择性达到100倍以上,正在进行一项I期临床试验,评估56在血液系统恶性肿瘤(AML,CLL,SLL和淋巴瘤)患者中的安全性和耐受性(NCT04588922)。
选择性CDK抑制剂的设计
CDK选择性抑制剂的设计需要考虑一些关键的结构特征。开发CDK1的选择性抑制剂,需要考虑配体与LYS89之间的静电相互作用。对于CDK2抑制剂,与GLU81和LEU83的氢键相互作用是保持活性的关键。由PHE80、VAL64、HIS84和GLN85组成的疏水口袋的存在有利于疏水相互作用和π-π堆积。对于CDK4/6的选择性抑制剂,需要与THR99/10产生极性相互作用,并且与保守铰链残基HIS92和VAL101的氢键相互作用也是必需的。对于CDK9抑制剂的开发,与ASP167和CYS106的强氢键相互作用十分必要。CDK9结合位点中的一些非保守区域,包括疏水区域和溶剂暴露区域,为设计选择性CDK9抑制剂提供可能。靶向CDK12的铰链区域,DFG基序和C端延伸区域是发现高活性和选择性CDK12抑制剂的可行方法。
总结
泛CDK抑制剂存在治疗窗窄的明显不足,选择性CDK抑制剂通过靶向特定的CDK亚型,安全性更高。但是由于不同CDK亚型的ATP结合口袋非常保守,因此发现ATP竞争性和高选择性CDK抑制剂面临重大挑战。选择性CDK抑制剂的发展依托于母核骨架和结构特征上的创新。目前,已经有四种CDK4/6抑制剂获批,其他的许多CDK抑制剂也处于临床研究阶段,具有巨大的应用潜力,期望未来在选择性CDK抑制剂的开发中取得更大的突破。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言