Erdheim-Chester病:症状体征、病因、流行病学、诊断与治疗
2022-08-17 MedSci原创 MedSci原创

Erdheim-Chester病(Erdheim-Chester Disease, ECD),又称脂质肉芽肿病或脂质肉芽肿瘤样增生症,是一种罕见的系统性非郎格汉细胞组织细胞增生性疾病,1930 年首次
Erdheim-Chester病(Erdheim-Chester Disease, ECD),又称脂质肉芽肿病或脂质肉芽肿瘤样增生症,是一种罕见的系统性非郎格汉细胞组织细胞增生性疾病,1930 年首次由 Jakob Erdheim和William Chester首次以“类脂质肉芽肿病”报道,1972年,Herry Jaffe再次报道该病时,将其命名为“Erdheim-Chester病”。该病主要以大量CD68(+) CD1a(-) 富含脂质的泡沫样组织细胞以黄色肉芽肿性增生浸润,伴有不同程度的纤维化和淋巴细胞浸润为特征,侵犯骨骼及多种器官。由于其形态学和免疫染色特点与幼年性黄色肉芽肿 (Juvenile xanthogranuloma, JXG) 相同,国际组织细胞协会将其纳入JXG疾病家族,近来研究认为ECD是不以皮肤侵犯为主的JXG特殊类型。它于2016年被世界卫生组织宣布为组织细胞肿瘤。
在2017年WHO分类中,将其单独列为一种疾病类型。ECD发病率极低,至今全世界报道约1000例,国内也有少数报道,属于罕见病。
2018年5月11日,ECD被列入国家卫生健康委员会等5部门联合制定的《第一批罕见病目录》。
一、一般概述
同义词(或曾用名):脂质肉芽肿病、多发硬化性组织细胞增生症
发病部位:常发生在长管状骨,一般为双侧及对称性分布,其他不常受累的部位包括肋骨、脊椎及颅面骨。中枢神经系统、心血管、肺、肾(腹膜后)和皮肤可受累。
二、症状与体征
ECD 导致称为组织细胞的免疫细胞过度产生,然后在体内的组织和器官中积累。可能涉及的身体部位包括长骨、腹膜后、皮肤、眼睛和眼睑、肺、脑、心脏、肾脏和垂体;然而,其他各种组织或器官也会受到影响。
ECD 的体征和症状因人而异,具体取决于具体位置和受累程度。例如,腿部的长骨通常会受到影响,导致骨痛。当眼睛周围的组织受到影响时,一个人的眼睑上可能会出现柔软的脂肪肿块或眼球突出。肺部受累可能导致肺纤维化,从而导致危及生命的并发症。垂体受累的人可能会发展为尿崩症,而大脑受累可能会导致各种神经系统症状。 ECD 患者可能出现的非特异性症状包括体重减轻、发烧、肌肉和关节疼痛以及不适。
ECD 的根本原因并不总是已知的。一些 ECD 患者的 BRAF 基因(最常见)或其他几个基因之一的获得性(非遗传)突变检测呈阳性,这可能使组织细胞无法控制地繁殖。该疾病的诊断依据是症状、显微镜下检查的相关组织活检(病理学)以及可能包括血液检查和影像学研究(如 X 射线、CT 扫描、MRI 和骨扫描)的专门测试。
1.骨:ECD骨累及是其主要特点,高达96%~100%,最常累及股骨远端、胫骨近端及远端,表现为双侧对称的骨干或干骺端骨硬化,但极少单累及骨,其中约50%出现骨痛症状,绝大多数骨受累的患者同时伴发骨外病灶。ECD患者骨质硬化双侧对称性分布在贯穿长骨的干骺端区域。86%的病例累及四肢长骨,最常受累的部位是股骨、胫骨和腓骨,少见于尺骨、桡骨和肱骨。骨痛常出现在膝和踝关节。除典型骨质硬化表现外,骨受累还可伴发骨骺部分硬化、骨膜炎和骨梗死。X线平片是检查ECD骨病变的主要手段,表现为特征性的四肢长骨对称性骨质硬化,偶有硬化和溶解病变混合存在,以下肢骨多见,但单纯溶骨性病变极其少见,可见骨髓质缺失,皮质不规则,骨膜增厚。CT、MRI和核素骨扫描对早期、隐匿的骨病灶更为灵敏。CT显示颅骨、脊柱、锁骨、肋骨等长骨之外的骨病变较平片更有优势。虽然以上骨受累的CT表现缺乏特异性,但当CT发现管状长骨、颅骨、胸骨、锁骨骨质硬化, 并伴发其他骨外病灶, 如脑膜占位性病变、胸主动脉"主动脉鞘"征象时, 高度提示ECD。核素骨扫描对早期骨病变灵敏度高,可见99mTc异常摄取,特征性表现为四肢长骨对称性高摄取骨显像剂99mTc-MDP,高度提示ECD [18]。MRI对ECD骨髓浸润灵敏性高,表现为正常髓腔内脂肪被异常增生组织所取代,呈不均一长T1、长T2信号,可见强化。除评价骨髓浸润程度和范围外,MRI还可清楚显示骨膜、软骨改变和骨梗死。
2.中枢神经系统 (CNS)及眶内间隙:ECD可累及下丘脑-垂体轴、脑实质、硬脑膜、脊髓、硬脊膜、眶内间隙等。CNS受累临床表现多样,病变的位置、大小和性质决定患者的临床表现,可有中枢性尿崩症、小脑性共济失调、全垂体机能减退、视神经水肿、突眼症等症状。CT和MRI是ECD累及CNS的主要检查方法。
下丘脑 - 垂体受累引起的尿崩症是 ECD 侵犯内分泌系统最常见的临床表现,89%的ECD患者存在尿崩症,78%的患者存在垂体前叶功能缺陷。半数ECD患者MRI检查可见下丘脑-垂体轴异常,常累及垂体柄和垂体后叶。垂体柄受累时表现为增粗或结节,呈明显均匀强化。Drier等报道下丘脑-垂体轴异常的ECD患者中,88%表现为垂体后叶的正常T1 WI高信号消失。
脑实质和硬脑膜也是ECD常累及部位。脑实质任何部位均可受累,最常累及幕下脑组织如小脑和脑干。小脑齿状核及邻近部位受累常表现为双侧对称性长T1、长T2信号,占位效应不明显,无明显强化。脑实质受累还可表现为结节或肿块,呈等T1、等或短T2信号,偶见长T2信号,呈明显均匀强化,周围无或伴轻中度水肿。脑膜病变常累及大脑镰、小脑幕及蝶鞍硬脑膜,表现为边界清晰的脑膜瘤样结节或肿块,也可表现为弥漫性硬脑膜增厚,T1、T2为等或低信号,呈明显均匀强化。其中19%的ECD患者可表现为脑实质和硬脑膜同时受累。有超过 25% 的病例可以单发或多发于硬脑膜,使硬脑膜局限性或弥漫性增厚。颅脑平扫 CT 检查可以发现脑实质病变呈低密度,硬膜病变多呈中度强化。CT对脑实质和硬脑膜ECD病变敏感性不及MRI,但可清晰显示颅骨骨质硬化。尽管CNS累及部位广泛,影像学表现缺乏特异性,但Drier等认为当颅内病变联合颅骨骨质硬化时可提示ECD。其通过对30例ECD患者的颅脑影像学研究发现,所有颅内病变均伴发颅骨骨质硬化或眶内球后占位。其中27%并发颅骨硬化和球后占位,23%并发颅骨硬化和硬脑膜病变,17%同时并发颅骨硬化、球后占位以及硬脑膜病变,10%同时并发颅骨硬化和垂体柄异常。
眶内间隙受累时可有突眼,眼周黄色瘤表现。MRI常表现为边缘清晰、包绕视神经的球后肿块或弥漫性球后脂肪浸润。
3.心血管系统:ECD累及心脏以心包、心肌受累为主,以心包侵犯较为常见。40%~45%患者心包受累,临床可表现为心包炎、心包填塞症状,CT表现为心包增厚和积液,增强扫描偶见强化。心肌侵犯时常见右心房受累,可表现为假瘤样肿块或弥漫性心肌肥厚,CT图像上病变密度类似心肌,MRI表现为T1、T2略低信号,增强扫描轻度均匀强化。右心房假瘤样肿块及弥漫性增厚可视为本病的特征性影像学表现。约20%的ECD患者存在心脏症状,心肌受累可引起瓣膜功能障碍和心电传导异常,极少数可以引起心衰,且与不良预后相关。
Haroche等研究发现,37例ECD中,75%的患者CT或MR发现心脏异常,49%累及右心,其中30%表现为右心房假瘤样肿块,19%冠状沟受累,14%心包增厚,24%心包积液。心包浸润是ECD最常见的心脏表现(约24%患者)。心肌受累的几率仅次于心包,主要表现为心肌肥厚,特别是右心房。不太常见的是,浸润影响心肌组织,尤其是右心房和瓣膜心内膜。ECD相关的ECG异常为PR段缩短,窦房阻滞,窦性心动过缓,二尖瓣型Q波异常,ST-T异常和轻度的ST升高。心血管系统受累的表现因病变位置和大小而不同。这些病变造成不同的临床表现:充血性心衰、心肌梗死、血栓栓塞、心脏重塑、瓣膜的功能障碍、缺血和外周水肿等。
约66%的ECD患者可见血管受累,主要侵犯主动脉及其分支,如左颈总动脉、腹腔干等。ECD累及静脉是很少见的。CT、MRI上可见血管被周围增生的软组织包绕,病变形态及累及范围多样,可对称或不对称,可闭合呈环形包绕或非环形,可包绕整大动脉或仅包绕某一段大动脉。Serratrice等将整段大动脉周围包绕称为"主动脉鞘征",为ECD的特征性表现。Haroche等报道66%的主动脉受累的患者中23%可见"主动脉鞘"征。血管周围浸润不局限于主动脉,ECD的血管受累通常无临床症状,但可能导致严重的临床后果,特别是当发生动脉狭窄时,包括由于颈动脉受累导致的脑缺血,由于冠状动脉受累导致的心肌梗死,肾动脉受累常导致肾性高血压,肠系膜上动脉受累引起的肠系膜缺血。
4.肺及胸膜:ECD肺受累较为常见,常见症状为隐匿进展数月或数年的干咳和呼吸困难。约53%患者累及肺实质,表现为不同程度的肺间质纤维化,是间质性肺病的一个原因,41%患者累及胸膜。ECD患者肺功能检查通常显示轻度限制性通气功能障碍,伴一氧化碳扩散能力正常或下降。动脉血气值通常是正常的。然而,随着疾病进展可能出现缺氧、高或低碳酸血症。ECD肺病变主要沿淋巴系统浸润,伴纤维化和淋巴、浆细胞炎性浸润,故影像常表现为间质性肺病的特征。高分辨率CT可评估肺或胸膜受累。肺部广泛浸润和纤维化可能导致严重的心肺症状,甚至心肺衰竭。HRCT为最佳检查方法,表现为肺间质纤维化征象,如小叶间隔增厚、小叶中心型微结节和磨玻璃影等,肺实变少见。胸膜受累时可见胸膜增厚和胸腔积液。当胸部CT发现上述征象并发胸骨、肋骨骨质硬化时,对ECD诊断有较高的提示意义。
5.腹膜后:大多数ECD累及腹膜后腔时常无临床症状,有时可伴腹痛、排尿困难,严重者可出现肾积水、肾性高血压、尿路梗阻甚至肾衰。当病变侵犯肾周脂肪和肾周筋膜时,CT、MRI表现为双侧对称的肾周软组织密度影,边界不规则,类似毛发状,呈特征性的"毛发肾"征(hairy kidney),约68%的患者CT检查可见"毛发肾"征,高度提示ECD诊断。当肾周和近端输尿管同时受累时,对本病的诊断具有更大的提示意义。肾周浸润可能进展至肾窦及导致肾后梗阻。肾周浸润和纤维化可能导致双边输尿管的阻塞导致肾积水和肾功能的下降,纤维化最易累及中段和远段输尿管。除肾脏外,肾上腺也可受累,表现为双侧肾上腺对称性增粗伴周围脂肪浸润,少数可表现为软组织肿块,通常无临床症状。Haroche等报道中仅1例(1/7)肾上腺累及患者有临床症状,表现为肾上腺素分泌不足。
6. 其它部位:ECD还可以累及其它部位,如皮肤、肌肉、乳腺、肝脏、脾、骨骼肌,一般表现为受累部位软组织肿块,相应功能受损的症状。
常规X线平片、骨扫描、CT和MRI只能对某些特定部位进行评价,部分影像学征象较为独特[18],如双侧对称的骨干或干骺端骨硬化, 四肢长骨对称性高摄取骨显像剂99mTc-MDP, "主动脉鞘征", 以及"毛发肾"征等。但进行整体性评估,难以满足临床全面把握病情的要求。全身PET能够同时评估ECD累及的病变部位最多。18F-FDG PET是近年来应用于临床的一种新型影像学检查方法,通过病变与正常组织糖代谢水平不同对病变进行检测,不但可以显示病变的数目、范围,还可反映病变活性。Arnaud等研究了PET在31例ECD患者中的应用价值,发现PET在不同部位(长骨、心脏、大血管、CNS、肺和胸膜、肾周间隙、副鼻窦、眶内间隙)初次诊断灵敏度相差迥异,最高为CNS(78.3%),最低为肾周间隙(4.3%),表明对ECD患者很多部位病变评估时,18F-FDG PET尚不能取代传统的影像学检查方法。
除PET外,全身MRI也可用于ECD的全身性评价,有研究证实其对心脏和CNS病变显示较好。但全身MRI需要高场强设备,检查耗时较长,且其在ECD诊断、病情评估和随访中的价值是否优于18F-FDG PET尚未证实。
实验室检查是非特异的,非诊断性的,可作为影像学和组织学的补充。目前对于ECD的诊断,需临床、影像和病理相结合。
三、病因
ECD中增生的非朗格汉斯细胞组织细胞可能来源于CD34+的骨髓前体细胞。目前,间质树突状细胞是假定起源细胞,但仍存在争论。迄今为止,ECD的病因及发病机制尚不明了,可能与基因突变、免疫失调和炎症有关。研究表明,相当一部分ECD和朗格汉斯细胞增生症 (Langerhans cell histiocytosis, LCH)患者存在BRAF基因突变,ECD与LCH可同时发生,提示两者关系密切或这些组织细胞之间存在相互转化。成人LCH和ECD患者骨髓祖细胞、单核细胞和髓系DC含有BRAFV600E等位基因。关于ECD是一种单克隆的肿瘤性疾病,还是多克隆的炎症反应性疾病这一问题仍然存在争议。ECD患者BRAF V600E突变表明该病是克隆性疾病,提示在ECD发病机制中决定性作用。这些突变的发现将导致组织细胞疾病LCH和ECD的新分类,如被归类为炎症性髓样肿瘤
自Blombery等首先证明ECD患者存在BRAF突变以来, 至今发现,有50% to 75% 的ECD患者存在该突变,中国ECD患者中突变率为68.8%。B-Raf是一个丝氨酸-苏氨酸蛋白激酶,参与Ras-Raf-Mek-Erk细胞分裂素活化蛋白激酶(mitogen-activated protein kinase,MAPK)转导通路。该信号通路通过细胞外生长因子结合到细胞膜酪氨酸激酶受体被激活,并调节细胞增殖和生存。BRAF V600E突变导致B-Raf的第600位谷氨酸替换为缬氨酸,导致丝氨酸/苏氨酸激酶构象改变,激活Ras-Raf-Mek-Erk途径,进而促进了细胞的增殖。使用BRAF抑制剂维罗非尼在该病中应用取得的积极疗效,也表明BRAF基因突变在疾病发生发展中具有重要作用。
一些ECD病例, 无BRAF V600E突变,但有MAPK和AKT通路的其他经常性体细胞突变,包括RAS和PIK3CA突变。表明,MAPK及AKT信号通路中的NRAS及PIK3CA突变也参与了ECD的发病过程, 进一步提示Ras-Raf-MeK-ErK通路在ECD发病中的重要作用。研究发现,约有 11 % ECD患者有 PIK3CA突变导致 mTOR(哺乳动物雷帕霉素靶蛋白) (PI3K/AKT/mTOR)通路的激活。mTOR通路由mTORC1和mTORC2两种不同的复合物参与。mTORC1对雷帕霉素很敏感,p70S6K的磷酸化增加说明 ECD中有mTORC1的参与。mTOR通过整合细胞内外的信号调节细胞的增殖、分化、凋亡和一些代谢过程,如:蛋白质和脂质的合成也受mTOR的调控。此外,mTOR能够通过促进B细胞、T细胞及抗原递呈细胞的分化和激活影响免疫过程。在肿瘤和炎症反应中可以发现 mTOR的异常激活。研究发现,在ECD中,mTOR显著磷酸化,与之一致的是mTOR的下游p70S6K的磷酸化也显著增加。在CD68 阳性的泡沫样组织细胞中mTOR和p70S6K的磷酸化显著增加。
ECD发病过程中存在一个明显、独特的促炎细胞/趋化因子网络,负责募集和激活组织细胞,进入ECD病变。研究表明,ECD患者IFN-α、IL-1β、IL-7、IL-12、TNF-α、IL-6和单核细胞趋化蛋白1(MCP1/CCL2)水平增加。ECD病变的组织细胞向炎症表型偏移,局部出现类似于Th1的可溶性细胞因子网络,如IL-1,IL-6,CCL2,CCL5,CXCL8,TNF-α,干扰素(IFN)-γ[14] [15],被称为“细胞因子风暴”。此外,细胞因子抑制剂,如IL-1受体拮抗剂anakinra, IL-6阻滞剂Tocilizumab,和TNF-α阻断药Infliximab治疗ECD患者取得了令人鼓舞的疗效,也证明细胞因子/趋化因子介导的组织细胞募集和激活的发病机制。
近来提出,由癌基因BRAF和RAS突变诱导的衰老(OIS) 可能作为ECD致病新的机制,与该病发生发展有关,这种机制本质上是疾病发展的原因。OIS是对抗以细胞周期阻滞和诱导促炎分子为特征的致癌事件的主要保护机制,作为致癌突变与观察到的炎症之间的可能联系,支持癌基因BRAFV600E突变在ECD发病机制中的核心作用。事实上,ECD在体内发生的变化涵盖了与OIS相关的分子事件,即细胞周期阻滞和强烈的局部炎症反应。因此,巨噬细胞对不同组织的浸润和ECD中观察到的炎性局部和全身效应可能代表OIS的不利之处。
四、鉴别诊断
1.播散型幼年性黄色肉芽肿(DJXD): 发生于10岁以下儿童,半数在1岁以内。与I 型神经纤维瘤病相关,无BRAF 突变。
2.Langerhans细胞组织细胞增多症(LCH):多见于儿童。肿瘤细胞呈现典型的Langerhans 细胞特征,有“咖啡豆样”核沟。背景较多嗜酸性粒细胞。表达 CD1a、S-100、Langerin,弱或不表达CD68和CD163,电镜下有Birbeck颗粒。
3.组织细胞肉瘤(HS):HS肿瘤细胞呈现明显异形性,细胞大,圆或椭圆形,胞质丰富、嗜酸性,核膜不规则。无泡沫状(脂质化)组织细胞,无Touton巨细胞。
4.Rosai-Dorfman 病:累及淋巴结或结外组织,表现为大量组织细胞增生。增生的组织细胞核圆形,较大,空泡状,核仁显著。胞质丰富,泡沫状,常含有完整的淋巴细胞和浆细胞。组织细胞免疫表型:S100+、CD68+。
5. Gaucher病:Gaucher细胞具有嗜酸性胞浆呈“皱纹纸”样,常与骨坏死相关,Gaucher病骨骼无硬化,骨髓梗死时可出现硬化。
五、诊断
要点:
1. 罕见,平均发病年龄55-60岁,也有发生于儿童的报道。病灶最常累及部位为骨骼(>95% )、心血管、腹膜后(肾)、中枢神经系统、肺、和皮 肤,并出现相应状。
2. 病变部位组织形态与免疫学与JXD相同:具泡沫状胞质(含脂)的组织细胞增生,常有Touton 巨细胞,并伴纤维化。可混有其它炎性细胞。
3. 组织细胞CD68+、CD163+、Factor VIIIa+、CD14+、S100- 、CD1a-、Langerin-。
4. X 线呈现特征性的对称性长骨干骺端硬化性改变。
5. 大多数病例(>50%)有BRAF(V600E)突变。此外,少数病例有NRAS突PI3KCA通道基因突变。
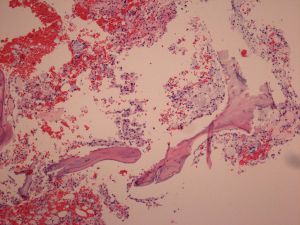
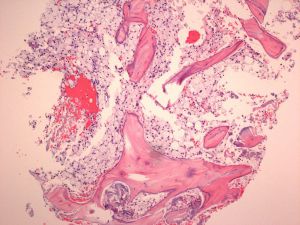
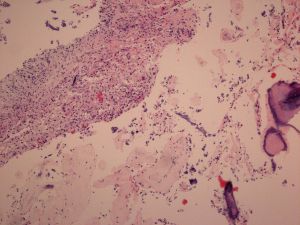
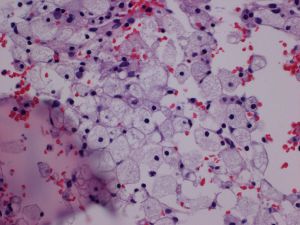
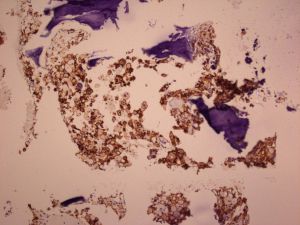
分子标记:
六、治疗
目前对于ECD的治疗尚存在争议。目前对于该病的治疗的方法主要包括糖皮质激素、环孢素、干扰素、化疗药物、手术切除、放疗等。因为病例数少, 对于疗效的观察数据不完善。
1. 基于ECD免疫的治疗,包括IL-1受体拮抗剂anakinra、IL-6阻滞剂tocilizumab,和TNF-α阻断剂英夫利昔(Infliximab)。
1). 干扰素-α是目前ECD的一线治疗,大多数患者可达到疾病稳定。目前已证明,干扰素α 对部分患者具有疗效,可以延长患者的生存时间。干扰素-α的疗效与疾病受累的部位有关。对治疗有反应的部位依次是皮肤,其次是中枢神经系统,垂体,肺和心脏。Hervier等用大剂量的干扰素治疗27例ECD患者,46%的患者症状得到改善,21%的患者病情稳定,总有效率为67%,被证实能延长患者总生存期。因干扰素-α需长期应用,故治疗耐受性是长期维持治疗的关键所在,并且在治疗心脏及CNS 受累的ECD中效果较差。因此,希望找到一种可以替代干扰素α对ECD进行有效治疗的方法。在这种情况下,用抗-TNFα单克隆抗体英夫利昔单抗和甲氨蝶呤的联合治疗似乎是有效且耐受良好的。靶向治疗 IL-1受体拮抗剂(Anakinra)取得了令人鼓舞的成果。
2). 克拉屈滨 (Cladribine,2-chlorodeoxyadenosine,2-CdA) 是一种用于干扰素治疗失败后活动性的伴有临床意义的贫血、中性粒细胞减少、血小板减少以及疾病相关症状的毛细胞白血病治疗药物。近来发现,该药物用于ECD治疗,可使患者症状显著好转,中枢神经系统病变达到部分缓解。克拉屈滨的副作用为剂量相关的骨髓抑制和神经毒性。有报道患者在克拉屈滨治疗过程中出现短暂失明,故克拉屈滨的使用过程中需密切监测眼球和视神经抑制的程度。
2. 基于基因突变的靶向治疗
1). 威罗菲尼(Vemurafenib)是第一个被FDA批准的BRAF V600E抑制剂通过抑制突变激酶活性,从而阻止这种突变细胞的增殖和诱导死亡。近年来发现,在ECD患者中有 50%~75% 患者出现了BRAFV600E突变,这使得人们在有关BRAF抑制剂治疗疗效带来希望。威罗菲尼治疗中存在这种突变的ECD患者疗效显著。多系统、难治性ECD的患者接受了BRAF抑制剂威罗菲尼治疗,使用此药治疗后疗效显著。伊马替尼或舒尼替尼抑制PDGF信号可产生一定疗效。索拉非尼(Sorafenib:多吉美, Nexavar),是一种新型多靶点抗肿瘤药物,具有双重的抗肿瘤作用,既可通过阻断由RAF/MEK/ERK介导的细胞信号传导通路而直接抑制肿瘤细胞的增殖,还可通过抑制VEGF和血小板衍生生长因子(PDGF)受体而阻断肿瘤新生血管的形成,间接地抑制肿瘤细胞的生长。
2). mTOR抑制剂雷帕霉素。雷帕霉素(Rapamycin,RAPA,RPM,又名 Sirolimus)及强的松在治疗ECD时能给患者带来的益处,为BRAF没有发生突变的ECD患者或者不能耐受干扰素α的患者带来希望。Gianfreda 及其同事对 mTOR的相关抑制剂进行了研究。治疗的过程中,患者总体耐受良好。尽管治疗的效果与BRAF突变的ECD患者接受 vemurafenib治疗的效果同样好,但该试验中,雷帕霉素及强的松的治疗效果确是不可忽视的。mTOR是细胞生长和增殖的重要调节因子,mTOR抑制剂具有抗增殖和免疫抑制的功能,在肾移植领域已被应用了很多年,目前也被应用到很多的疾病如:恶性肿瘤及同种异体移植排斥的治疗过程。
3. 其它治疗还包括糖皮质激素,阿那白滞素、细胞毒性药物,例如长春新碱、环磷酰胺,阿霉素等。不同类型的化疗方案虽取得了不同程度的疗效,但大多数情况下缓解非常短暂。局部放疗曾用于治疗眼眶病变。外科手术为特定的情况下的一个临时解决方案。尽管迄今为止采用了几种治疗策略,但预后仍然很差。
七、ECD评估
ECD发病率低,缺乏足够的样本量进行分析,目前对于ECD的影像学检查方法与策略选择主要基于个案报道、小样本研究以及临床经验。ECD确诊后,定期、全面的复查尤为重要。ECD患者应每3个月复查局部CT或MRI,病情平稳后可每6个月复查。并且,ECD患者应每3~6个月复查FDG-PET。继续密切地定期随访本例患者局部和全身情况,如出现其他系统累及应给予治疗干预。
2014年ECD全球联盟(http://www.Erdheim-Chester.org)聚集ECD研究领域内多学科专家,共同出台了首个ECD专家共识性诊治指南,对于ECD的诊断、疗效评估及随访中的影像学检查选择给出了如下指导意见:
1.病情的评估:所有患者需做全身PET/CT(含四肢)、胸腹盆CT、颅脑增强MRI、鞍区MRI、心脏MRI。有局部受累症状的患者可加做眼眶MRI增强扫描、肾动脉超声、胸部HRCT、肺功能检查、睾丸超声、心电图等。
2.疗效观察,预后及随访:治疗后每隔3~6个月行全身PET/CT检查,病情稳定后检查间隔可适当延长;治疗开始后每隔3个月行受累部位特异性的影像学检查(如胸部行CT扫描、CNS行MRI检查等)。病情稳定前后, 相应临床症状改善或者专科检查(如肾功能检查)显示病情改善者可改为每隔6个月复查。
八、ECD预后:
为慢性临床过程,预后取决于疾病范围。中枢神经系统或多器官系统累及患者预后不良。
ECD的预后较LCH差, 主要与内脏浸润程度有关,中枢神经系统、心血管和肺脏累及治疗效果不佳,分别是预后的独立风险因素。呼吸窘迫、广泛的肺组织纤维化、心衰是主要的死亡原因。3年生存率为50%,尚无自行缓解的报道。目前,ECD的预后预测是非常谨慎。Veyssier-Belot等中位随访37例ECD患者2.7年,22例(59%)患者在随访期内死亡,8例患者在诊断后6个月内死亡。Arnaud等报道的1年和5年生存率分别为96%和68%。由于临床病程和预后受疾病受累部位的影响很大,因此ECD疗程范围从无症状到危及生命,据报道全球5年死亡率为30-40%。
九、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
https://rarediseases.info.nih.gov/diseases/6369/erdheim-chester-disease
杨守京,Erdheim-Chester病的诊治及其研究进展 https://blog.sciencenet.cn/blog-548274-1192032.html
https://rarediseases.org/gard-rare-disease/erdheim-chester-disease/
Chester W. Über lipoidgranulomatose. Virchows Arch Pathol Anat Physiol. 1930; 279(2):561-602.
Brousse N, Pileri SA, Haroche J, Dagna L, Jaffe R, Fletcher CDM, et al. Erdheim-Chester disease. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., eds. World Health Organization Classification: Tumours of Hematopoetic and Lymphoid Tissues. Revised 4th ed. Lyon, France: IARC Press; 2017:481-482.
Milne P, Bigley V, Bacon CM, Neel A, McGovern N, Bomken S, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017; 130(2):167-175.
Haroche J, Cohen-Aubart F, Charlotte F, Maksud P, Grenier PA, Cluzel P, et al. The histiocytosis Erdheim-Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol. 2015; 11(9):1033-1042.
Blombery P, Wong SQ, Lade S, Prince HM. Erdheim-Chester disease harboring the BRAF V600E mutation. J Clin Oncol. 2012; 30(32):e331-332.
Haroche J, Charlotte F, Arnaud L, von Deimling A, Helias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012; 120(13):2700-2703.
Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013; 121(9):1495-1500.
Haroche J, Cohen-Aubart F, Emile JF, Maksud P, Drier A, Toledano D, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015; 33(5):411-418.
Emile JF, Diamond EL, Helias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014; 124(19):3016-3019.
Cao XX, Sun J, Li J, Zhong DR, Niu N, Duan MH, et al. Evaluation of clinicopathologic characteristics and the BRAF V600E mutation in Erdheim-Chester disease among Chinese adults. Ann Hematol. 2016; 95(5):745-750.
Diamond EL, Abdel-Wahab O, Pentsova E, Borsu L, Chiu A, Teruya-Feldstein J, et al. Detection of an NRAS mutation in Erdheim-Chester disease. Blood. 2013; 122(6):1089-1091.
Xu Y, Li N, Xiang R, Sun P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends in biochemical sciences. 2014; 39(6):268-276.
Haroche J, Amoura Z. mTOR: a new target in Erdheim-Chester disease? Blood. 2015; 126(10):1151-1152.
Arnaud L, Gorochov G, Charlotte F, Lvovschi V, Parizot C, Larsen M, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011; 117(10):2783-2790.
Munoz J, Janku F, Cohen PR, Kurzrock R. Erdheim-Chester disease: characteristics and management. Mayo Clin Proc. 2014; 89(7):985-996.
Cavalli G, Biavasco R, Borgiani B, Dagna L. Oncogene-induced senescence as a new mechanism of disease: the paradigm of erdheim-chester disease. Front Immunol. 2014; 5:281.
Cangi MG, Biavasco R, Cavalli G, Grassini G, Dal-Cin E, Campochiaro C, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis. 2014.
Diamond EL, Dagna L, Hyman DM, Cavalli G, Janku F, Estrada-Veras J, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014; 124(4):483-492.
Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016; 127(22):2672-2681.
Diamond EL, Subbiah V, Lockhart AC, Blay JY, Puzanov I, Chau I, et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data From the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA oncology. 2018; 4(3):384-388.
Gianfreda D, Nicastro M, Galetti M, Alberici F, Corradi D, Becchi G, et al. Sirolimus plus prednisone for Erdheim-Chester disease: an open-label trial. Blood. 2015; 126(10):1163-1171.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#诊断与治疗#
38
#Chest#
54
#EST#
39
#流行病#
0