NEJM:真实世界证据,路在何方?
2022-05-09 NEJM医学前沿 NEJM医学前沿

距离2016年通过《21世纪治愈法案》(21st Century Cures Act)已过去5年多,“真实世界数据”(RWD)和“真实世界证据”(RWE
距离2016年通过《21世纪治愈法案》(21st Century Cures Act)已过去5年多,“真实世界数据”(RWD)和“真实世界证据”(RWE)两个术语的用法仍然不统一,而且有时两者还在互换使用。由于使用方面的不精确,我们在评估上述数据和证据产生的影响时,工作变得复杂,并且妨碍我们追踪其使用情况。
美国食品药品管理局(FDA)在其《FDA真实世界证据计划框架》(Framework for FDA's Real-World Evidence Program)中将RWD定义为“从各种来源常规收集的、与患者健康状况和(或)医疗服务相关的数据”,将RWE定义为“通过分析RWD得出的、关于医疗产品使用以及潜在获益或风险的临床证据”;这两个定义不考虑研究设计的类型。
但关于上述术语有两个普遍误解。第一个误解是认为RWD和RWE是2016年提出的全新概念。事实上,数据来源和研究设计类型始终未发生根本变化,但我们通过电子方式获取更详细临床数据的能力不断发展,而且此类信息的可靠性及与研究的相关性不断提高。此外,关于影响健康结局的临床因素,我们如今可以获得更为稳健的数据,因而有机会探索可替代随机化的各种统计学方法。然而,在研究概述中包含“RWD”或“RWE”并不能告诉我们数据究竟来自哪里,也不能告诉我们采用的是哪种研究架构。提供更多关于数据来源和研究设计的细节可减少对RWD和RWE的混淆。
第二个误解是认为随机对照试验(RCT)和观察性研究这一简单二分法可以描绘研究设计的全貌。随机分配治疗方案是RCT的关键优势,但并非所有临床试验都是随机试验;事实上,临床试验的关键特征是根据研究方案分配治疗。例如,在单组试验中,研究人员分配参与者接受干预措施,而不经过随机化,此类试验与观察性研究面临类似的挑战,即难以确定根据研究方案设立的研究组和对照组之间的临床结局差异是否代表实际治疗效果。
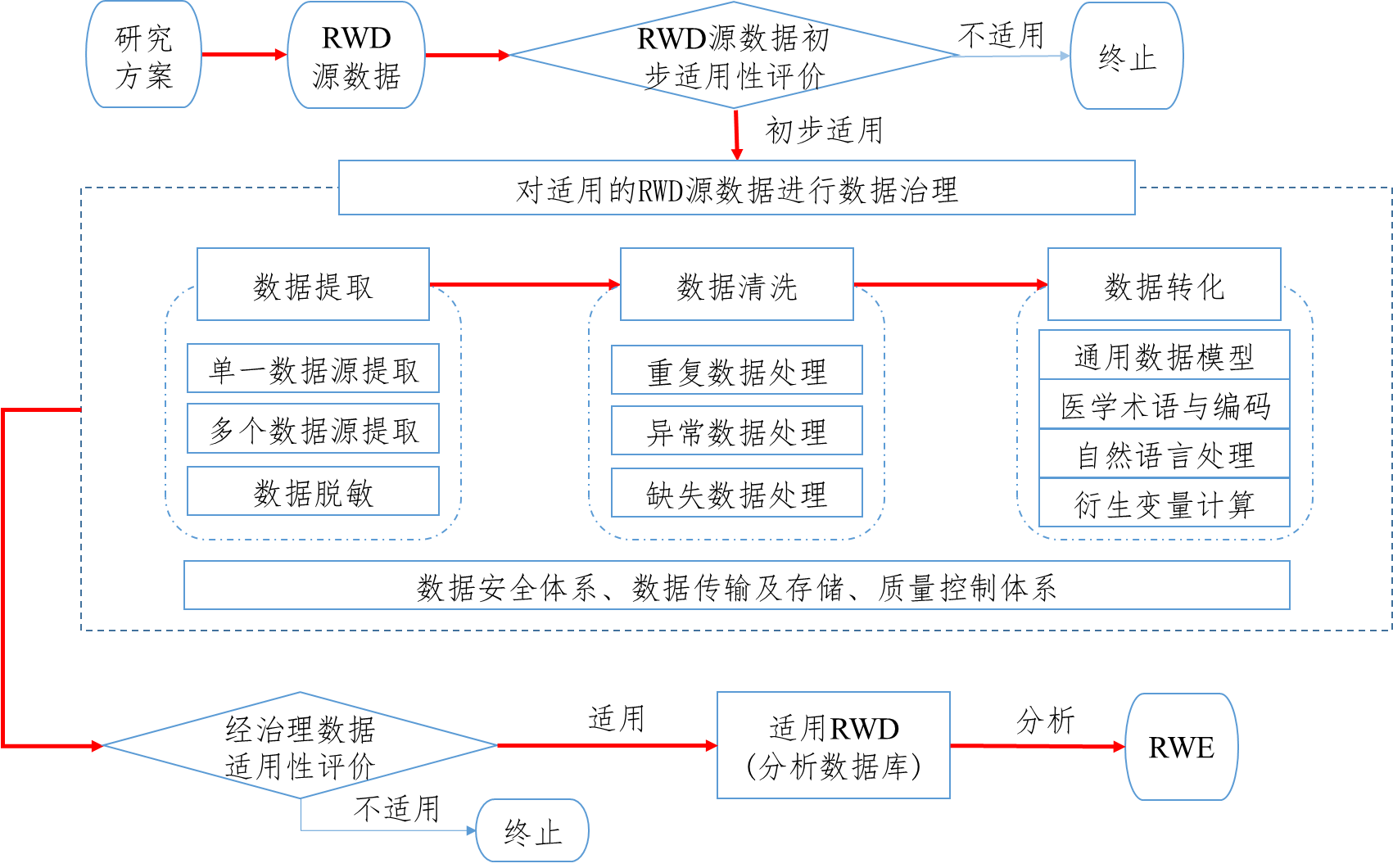
要纠正以上两个误解,需要认识到对RWD的依赖程度因研究设计类型的不同而异,并且认识到根据定义,纳入RWD的RCT会产生RWE(图1)。这一概念指出,即使严格的纳入标准已限制试验结果的普遍适用性,试验参与者仍然是存在于“真实世界”中,其结局也是发生于“真实世界”中,尽管人们普遍认为RWE都是在临床试验之外产生的。此外,“临床试验”和“观察性研究”两个术语在正确使用时具有明确含义,而“干预性研究”和“非干预性研究”两个术语的优势是可以说明关注的治疗方法是否是按照研究方案施用的。

图1. 各类研究设计对RWD的依赖程度
RCT表示随机对照试验;RWD表示真实世界数据;RWE表示真实世界证据。
如果有关治疗效果的因果推断主要依赖于干预性研究,并且在传统RCT中收集原始数据,则上述概念上的区别不太重要。然而,RCT越来越多地纳入RWD,而且当随机化因伦理或其他原因而不可行时,外部对照试验包括了完全来自二次数据来源(治疗组的“外部”)的对照组。另一方面,对登记系统收集的原始数据进行分析的非干预性研究开展得越来越多。
尽管人们对于这些术语和概念存在混淆,但在FDA工作的我们会在考虑监管决策时继续评估RWD和RWE。事实上,本机构在2021年发布了四份相关的指南文件草案。对于来自电子健康记录和医疗理赔数据库的数据,FDA指南包括以下几方面建议:如何选择相关数据来源,以及如何定义和验证研究变量;其他指南中的建议则涉及如何设计或使用现有登记系统,从而支持监管决策。关于数据标准的指南文件建议申办方记录变更理由,以确保RWD符合FDA支持的数据标准;关于监管考虑因素的指南文件说明了FDA对仅使用RWD的非干预性(观察性)研究的期望。
数字医疗技术产生的数据如果是在临床试验的背景下提供,则不符合RWD的严格定义,但它们在临床研究中的适用性值得一提。此类技术(包括用于远程获取生理或行为数据的软件应用程序和传感器硬件)在医疗领域发挥的作用越来越大,如果它们产生的数据经过验证并且有效,将为药物研发提供巨大机会。
支持在药物研发中应用RWD和RWE的FDA其他项目包括各种示范项目,它们的目的是提高RWD的实用性,探索可产生RWE的研究设计方法和数据分析方法,或者开发可推动上述过程的特定工具和技术。一个例子是OneSource项目(www.fda.gov/science-research/advancing-regulatory-science/source-data-capture-electronic-health-records-ehrs-using-standardized-clinical-research-data),该项目正在开发使结构化数据从电子健康记录自动流入外部系统的方法,目的是推动研究并且缩小患者医疗和临床研究之间的距离。
《21世纪治愈法案》通过之前,我们已经在依据现在所称的RWE批准药物和生物制剂,但过去5年间的两项批准可以阐明我们在本文中提出的问题。2021年,药品评价和研究中心(Center for Drug Evaluation and Research,CDER)批准他克莫司(普乐可复)联合其他免疫抑制剂,用于预防肺移植患者的器官排斥反应(www.fda.gov/drugs/news-events-human-drugs/fda-approves-new-use-transplant-drug-based-real-world-evidence),它依据的是在完善的登记系统和历史对照之间进行数据比较的非干预性究。用于肺移植患者是他克莫司的新适应证,该适应证获得FDA批准依据的是RWE,而且与患者和临床医师的期望一致,此外,它还代表CDER首次接受了“观察性研究”,并将其作为充分且有良好对照的研究,为有关治疗效果的确实证据提供主要支持。
2019年,生物制品评价和研究中心(Center for Biologics Evaluation and Research)批准了基于腺相关病毒载体的基因疗法onasemnogene abeparvovec-xioi(Zolgensma),将其用于携带特定双等位基因突变的2岁以下脊髓性肌萎缩患者(www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease)。该疗法获得批准的依据是对单组试验中接受该生物制剂的参与者主要结局(存活并达到功能里程碑)所做的评估,以及对这些结局与该疾病自然史研究中患者RWD所做的比较。虽然RWD在这个例子中的作用不及在他克莫司获批中那么突出,但在上述两个例子中,审查人员均发现数据适用,并且得出结论认为研究设计解决了监管机构关注的问题,而且研究实施过程符合FDA要求。
CDER批准他克莫司也凸显出观察性设计并不等同于使用二次数据。如果原始数据是在非干预性研究中收集(例如使用登记系统开展并以标准化格式收集数据的研究),则在临床评估方法和评估时机的差异方面,研究者遇到的挑战可能比使用电子健康记录、医疗理赔数据库或其他数据来源时遇到的挑战要小。在数据质量方面,更为一般性的问题包括临床相关性和可靠性(例如准确性、完整性、来源和可追溯性)。
其他考虑因素包括COVID-19疫情是否以及如何改变了人们对RWD和RWE以及相关实践的看法。一般而言,疫情加速了人们对RWD和RWE的认识和采纳,但它们的应用在疫情发生之前已经在逐渐增多。此外,尽管稳健RWE有时指导了疫情应对,但诊断、治疗和报告新疾病所涉及的挑战可能会产生方法学问题(研究可能因其他原因而无效,例如学术不端)。总的来说,COVID-19提供了运用RWD指导临床和监管决策的机会,但必须保持科学的严谨性。
FDA将继续致力于制定符合《21世纪治愈法案》的文件政策,同时维护证据标准,以履行我们保护和促进公众健康的责任。关注干预性研究和非干预性研究之间的区别可帮助研究者、申办方和监管机构更好地理解和描述相关方法学问题。积累更多经验,包括进行严格的非干预性研究,将有助于推进药物研发。
参考文献
Concato J and Corrigan-Curay J. Real-world evidence — where are we now? N Engl J Med 2022; 386:1680-1682.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#真实世界#
46
#真实世界证据#
42
NEJM上果然牛,感谢梅斯更新及时
33