
从皮肤科医生角度看血管炎
2023-08-02 海龙话皮 海龙话皮 发表于上海

皮肤是人体最大的组织器官和重要的免疫器官,一方面,皮肤是免疫反应的效应器官,同时也是免疫反应攻击的靶组织,因此,皮肤是发生血管炎最常见的靶器官。
血管炎是指组织病理学上血管壁及其周围组织的炎性改变,严重者可有血栓形成、甚至整个血管的破坏,导致出血性和/或缺血性病变,最终发生组织坏死或梗死。血管炎可发生于人体任何一种组织器官,动脉和静脉均可受累。根据血管炎累及的范围可分为单器官血管炎(局限于某一器官)和系统性血管炎(伴多系统损害)。皮肤是人体最大的组织器官和重要的免疫器官,一方面,皮肤是免疫反应的效应器官,同时也是免疫反应攻击的靶组织,因此,皮肤是发生血管炎最常见的靶器官。
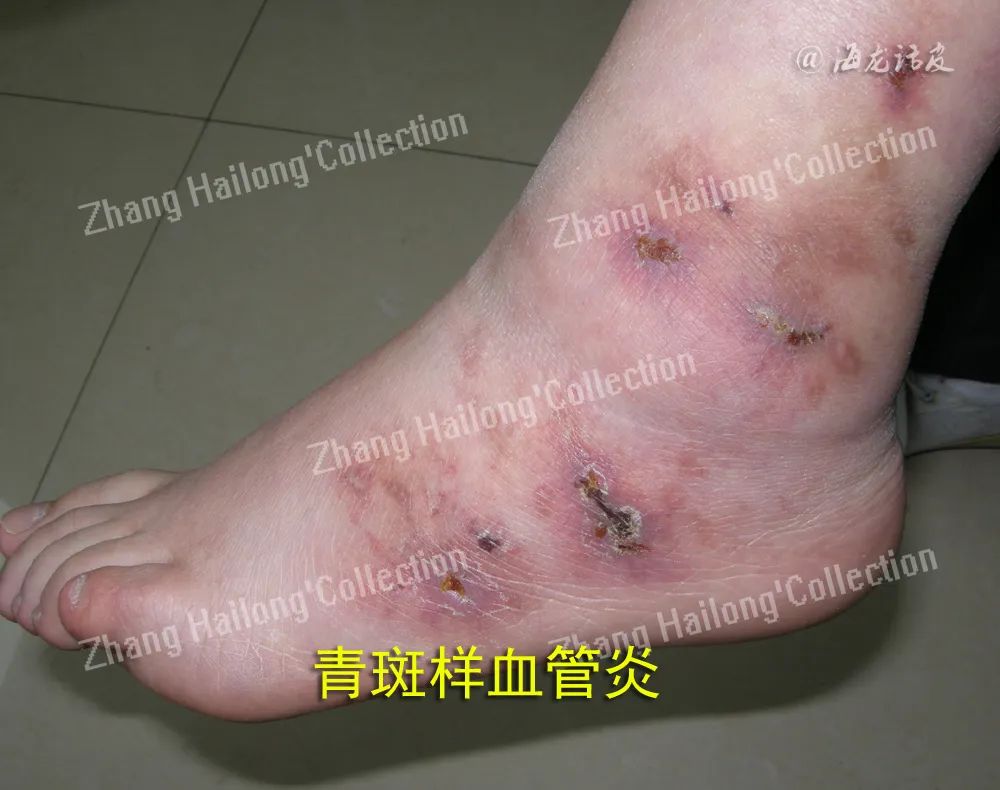
皮肤血管炎多见于以下情况:(1)原发性皮肤血管炎(仅累及皮肤而无系统损害),如变应性皮肤血管炎;(2)皮肤血管炎伴系统损害,如过敏性紫癜(也称IgA血管炎);(3)系统性血管炎的皮肤表现,如抗中性粒细胞胞浆抗体(ANCA)相关性血管炎。
皮肤血管炎的病因及发病机制
皮肤血管炎在临床上并不少见,研究结果显示年发病率39.6/百万~59.8/百万,另有研究发现年发病率为15.4/百万~29.7/百万,女性稍多,成人多于儿童(过敏性紫癜和婴幼儿急性出血性水肿除外),平均发病年龄约47岁。皮肤血管炎可能的病因包括感染(乙型肝炎病毒、单纯疱疹病毒、细小病毒B19、链球菌、葡萄球菌、分枝杆菌等)、药物(抗生素、抗癫痫药、非甾体类消炎药、利尿剂等)、系统性炎性疾病(结缔组织病、炎症性肠病、冷球蛋白血症等)和肿瘤(主要是血液系统肿瘤和实体瘤)。
大部分皮肤血管炎由免疫机制介导,涉及到4种类型的超敏反应。不同的超敏反应所引起的血管炎类型和临床表现不一,如Ⅰ型超敏反应通过嗜酸性粒细胞及IgE引起嗜酸性血管炎;Ⅱ型超敏反应由自身抗体(如抗内皮细胞抗体、ANCA)介导,引起血栓性血管病变及淋巴细胞性血管炎;免疫复合物介导的Ⅲ型超敏反应(Arthus反应)引起白细胞碎裂性血管炎;Ⅳ型超敏反应主要诱导肉芽肿性浸润,引起肉芽肿性血管炎。此外,个体差异、血流改变、慢性炎症反应、高凝状态等均可影响血管炎的病情发展和临床表现。
皮肤血管炎的临床和病理表现
皮肤血管炎的病程可表现为3种模式:(1)急性自限性:皮损常在6个月内消退,多与药物或感染相关,常见于变应性皮肤血管炎;(2)反复发作性:常见于过敏性紫癜和冷球蛋白血症性血管炎;(3)慢性持续性:常见于原发性系统性血管炎、结缔组织病相关性血管炎及副肿瘤性血管炎。
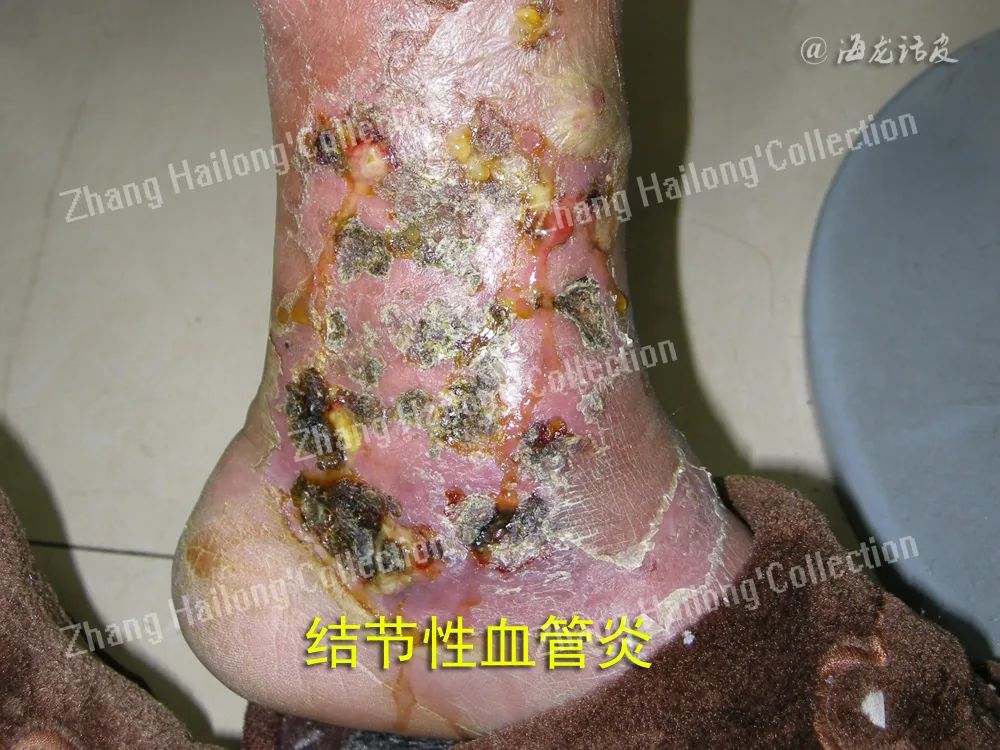
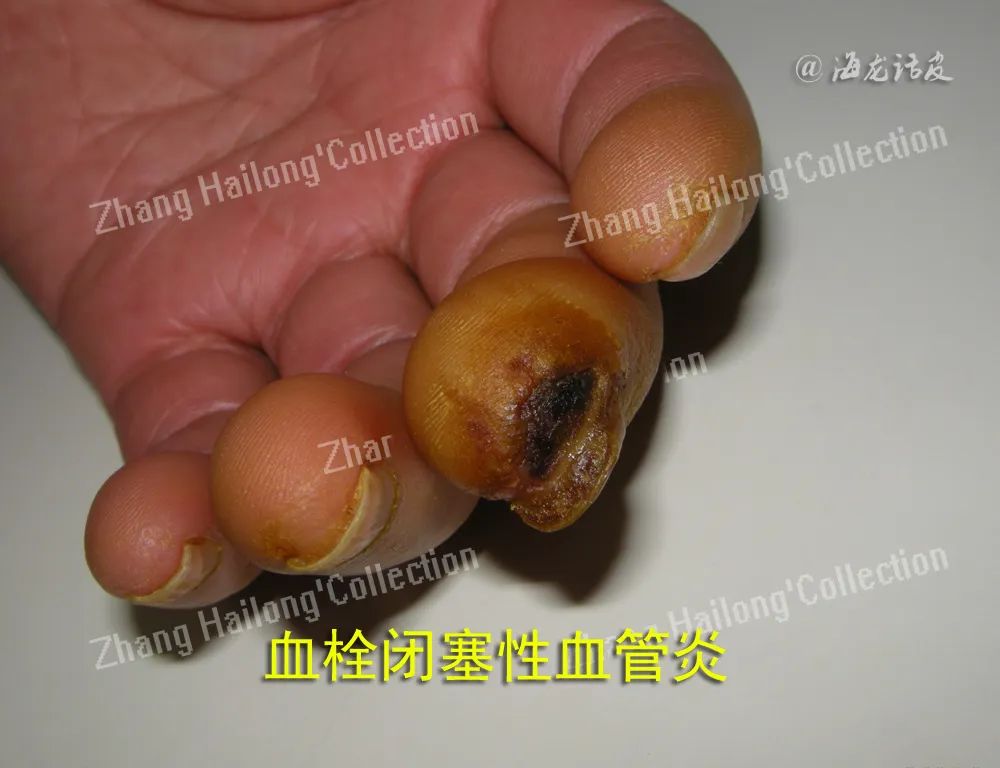
皮肤血管炎按受累血管部位可分为小血管炎、肌性血管炎和混合性血管炎。小血管炎累及非肌性血管如毛细血管、毛细血管后微静脉和微静脉,主要表现为可触及性紫癜、红斑、风团、丘疹、水疱/大疱、血疱、结节、坏死等,见于过敏性紫癜、变应性皮肤血管炎、荨麻疹性血管炎等。肌性血管炎累及肌性血管,表现为皮下结节、斑块、溃疡、指/趾端坏疽、网状青斑等,见于结节性多动脉炎、浅表血栓性静脉炎、硬红斑等。
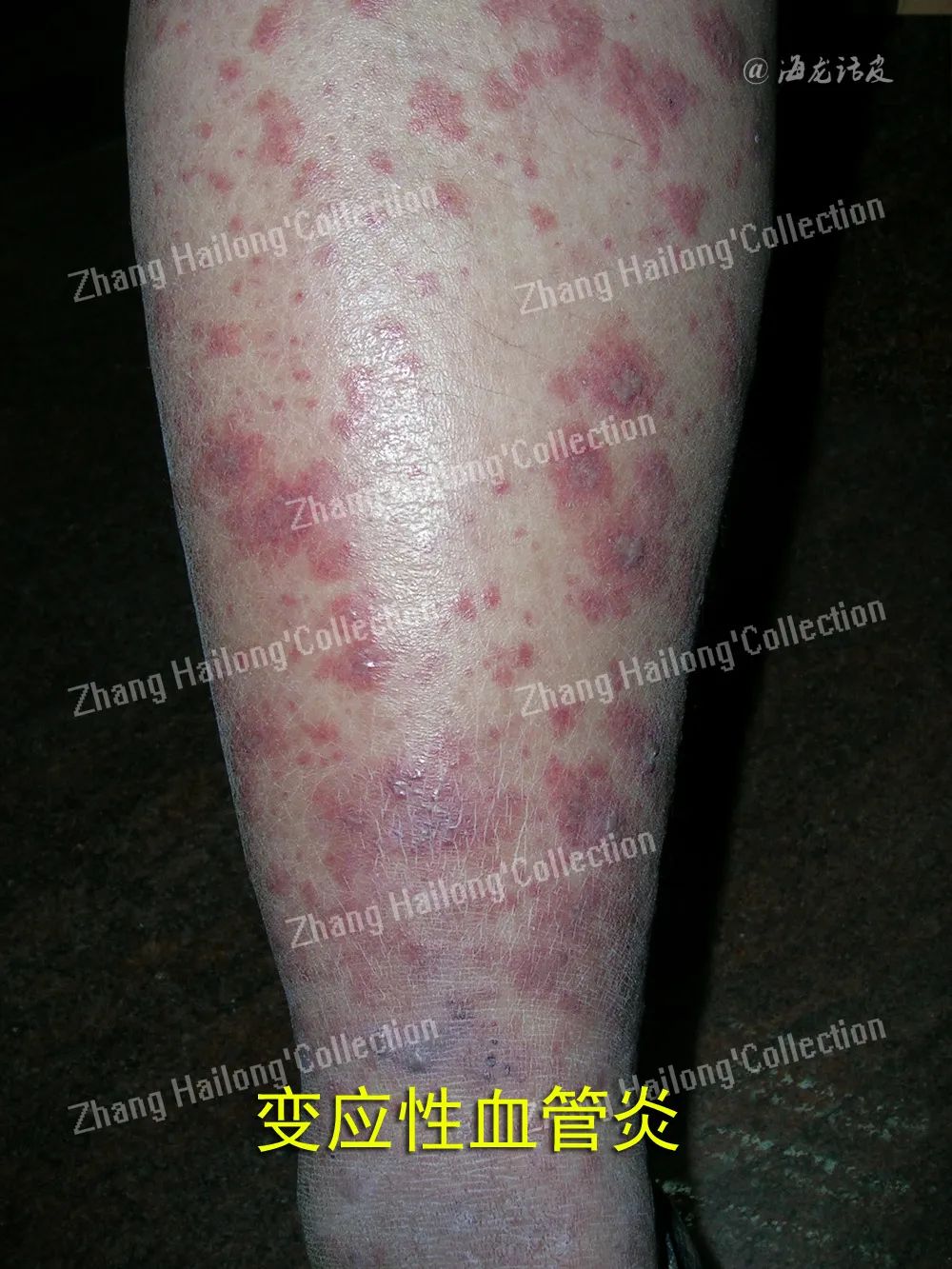
混合性血管炎既可累及非肌性血管,也可累及肌性血管,临床表现更具多样性,兼有小血管炎和肌性血管炎的临床表现,见于系统性血管炎如Wegener肉芽肿(又称肉芽肿性多血管炎)、Churg⁃Strauss综合征(又称嗜酸性肉芽肿性多血管炎)或显微镜下多血管炎等。
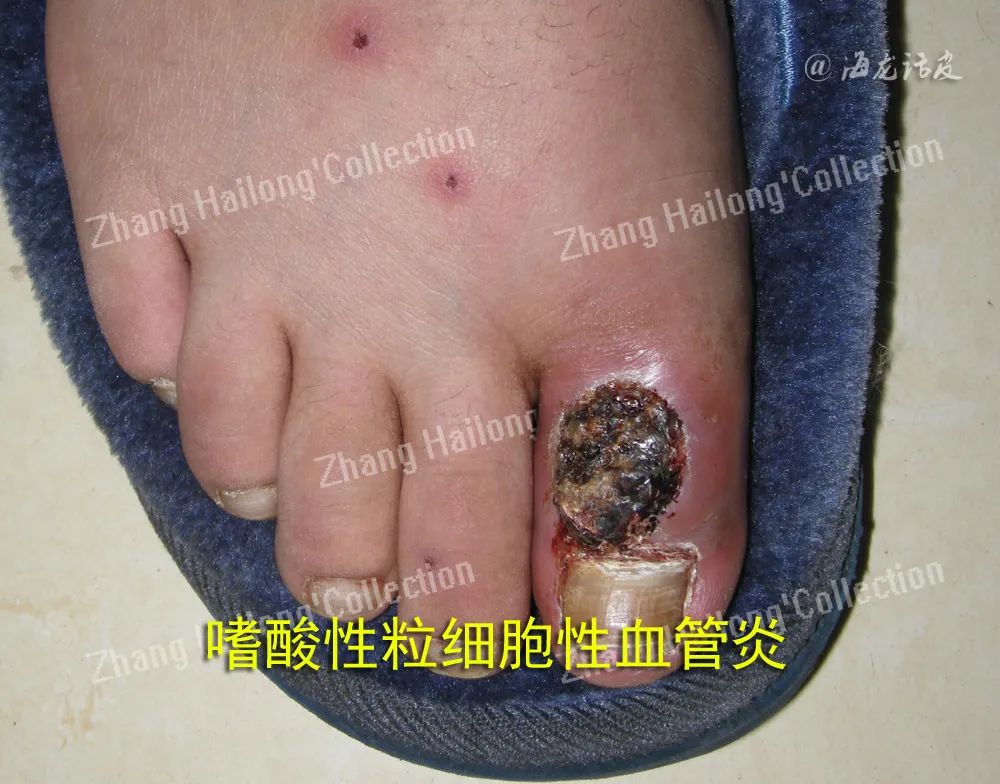
皮肤血管炎易发于静脉血流淤滞部位如双下肢,一般无明显自觉症状,可有烧灼感或轻微疼痛、 瘙痒。对于发生于双下肢的出血性皮损,应考虑皮肤血管炎的可能。如果皮损累及躯干及上肢,合并发热、关节痛、肌痛、食欲减退等系统症状,并伴其他皮外组织器官的症状,应考虑系统性血管炎或继发性血管炎(如结缔组织病相关性血管炎或类风湿性血管炎)。
组织病理是皮肤血管炎诊断的金标准。皮肤血管炎的组织病理特点:血管周围和/或血管壁炎性细胞浸润、炎性细胞破坏血管壁;血管壁和/或血管腔纤维蛋白沉积(纤维蛋白样变性);红细胞外溢;血管周围核尘(白细胞碎裂);内皮细胞肿胀、坏死、脱落。根据浸润炎性细胞的不同,皮肤血管炎又可分为白细胞碎裂性血管炎、淋巴细胞性血管炎、嗜酸性血管炎和肉芽肿性血管炎。
需要注意的是,如果组织中出现大量嗜酸性粒细胞,是诊断药物诱导血管炎的线索。如果血管炎累及真皮全层和皮下脂肪、不同大小血管及动静脉同时受累,出现栅栏状嗜中性肉芽肿性皮炎时, 应考虑系统性血管炎。
皮肤血管炎的诊断和鉴别诊断
皮肤血管炎一般根据病史、临床表现、组织病理学特征多可确诊。辅助检查包括血常规、尿常规、血凝全套、免疫学(抗核抗体/抗可溶性抗原抗体、ANCA、免疫球蛋白、补体、类风湿因子、抗磷脂抗体、冷球蛋白)、血液生化(肝肾功能、C-反应蛋白)、微生物学、肿瘤标志物(60岁以上老人)、胸部X线等检查,以了解内脏损害情况及排除系统性血管炎。
中等大血管炎和大血管炎需做血管造影。因皮肤血管炎表现的多样性,容易和一些非血管炎性疾病混淆,包括皮肤出血(血小板减少性紫癜、日光性/老年性紫癜、色素性紫癜样皮病等)、栓塞(胆固醇栓子)及血栓形成(青斑样血管病、暴发性紫癜、蛋白C/蛋白S缺乏、系统性淀粉样变病等),应注意鉴别诊断。
常伴胃肠道疾病的皮肤血管炎
皮肤病常与胃肠道疾病密切相关,部分皮肤血管炎可伴有胃肠道病变,甚至可为某些胃肠道疾病 的诊断提供警示。
炎症性肠病(inflammatory bowel disease,IBD)可伴发皮肤血管炎,包括白细胞碎裂性血管炎、结节性多动脉炎、肉芽肿性小血管炎、IgA血管炎、结节性血管炎、荨麻疹性血管炎、淋巴细胞性血管炎、ANCA相关性血管炎(肉芽肿性多血管炎、嗜酸性肉芽肿性多血管炎和显微镜下多血管炎),经治疗后皮损可消退。抗肿瘤坏死因子(tumor necrosis factor,TNF)-α生物制剂也可诱发皮肤血管炎。克罗恩病可伴发持久性隆起性红斑和坏疽性脓皮病。
过敏性紫癜的皮肤损害为可触性紫癜,常伴关节痛、肾脏损害及胃肠道病变。胃肠道症状多为非 特异性,与其他胃肠道疾病的症状类似,可表现为腹痛、恶心、呕吐、黑便和便血,严重者可出现消化道大出血、肠套叠、肠梗阻、肠梗死和穿孔,少数患者可发展为蛋白丢失性肠病。内镜显示黏膜充血、红斑、糜烂、溃疡、血管扩张等。组织病理提示白细胞碎裂性血管炎。
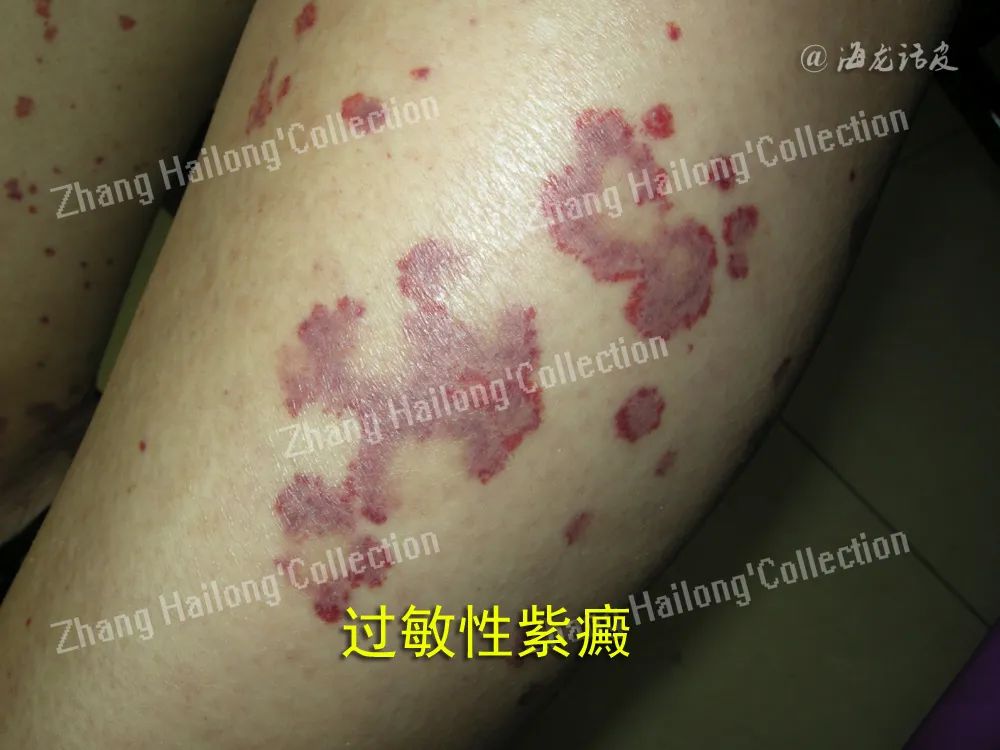
结节性多动脉炎是一种既可累及皮肤,也可累及内脏的肌性血管炎,14%~65%的患者可有胃肠道受累,特别是小肠。餐后腹痛是最常见的表现, 呈间歇性,其他症状包括恶心、呕吐、黑便、便血、腹泻或便秘。如缺血仅限于黏膜或黏膜下层,可发生溃疡和出血;如出现贯壁性缺血,则可出现肠壁坏死和穿孔,预后不良。
IBD或经历肠外科手术的患者可出现肠病相关性皮肤病-关节炎综合征。这是一种伴皮肤损害的嗜中性皮病,表现为皮肤红斑、丘疹、水疱、脓疱;组织病理表现为脓疱性血管炎(表皮内脓疱伴白细胞碎裂性血管炎)。
恶性萎缩性丘疹(Degos病)是一种罕见的闭塞性血管病(曾归类在淋巴细胞性小血管炎中),可累及皮肤、胃肠道、中枢神经系统等多个组织器官。皮损常为首要表现,表现为淡红色丘疹、坏死,形成特征性中央凹陷的瓷白色萎缩斑。胃肠道病变多见于小肠,表现为腹痛、腹胀、腹泻或便秘,重者小肠梗死、穿孔,导致腹膜炎。
皮肤血管炎的治疗
原发性皮肤血管炎:
轻症患者使用非甾体类消炎药、秋水仙碱、氨苯砜、沙利度胺即可获得满意 疗效;中、重度患者推荐糖皮质激素单一治疗;对于顽固性、糖皮质激素抵抗者可联合免疫抑制剂如甲氨蝶呤、硫唑嘌呤、环磷酰胺或环孢素A。
伴系统损害的皮肤血管炎:
推荐糖皮质激素联合环磷酰胺作为诱导缓解期的治疗,维持期可选择甲氨蝶呤、吗替麦考酚酯(如难治性低补体血症荨麻疹性血管炎、紫癜性肾炎)、硫唑嘌呤或环孢素A(如严重紫癜性肾炎)。治疗抵抗的难治性内脏血管炎可考虑生物制剂如英夫利西单克隆抗体或利妥昔单克隆抗体。
系统性血管炎(ANCA相关性血管炎):
ANCA相关性血管炎在皮肤科比较少见,预后相对较差。欧洲抗风湿病联盟(EULAR)的推荐方案:无内脏损害者,采用甲氨蝶呤或吗替麦考酚酯联合糖皮质激素治疗;有内脏受累或危及生命者,采用环磷酰胺或利妥昔单克隆抗体联合糖皮质激素(6个月)治疗;急进性肾衰竭或肺出血者,进行血浆置换;缓解期患者,采用硫唑嘌呤或甲氨蝶呤或利妥昔单克隆抗体维持治疗,糖皮质激素逐渐减量,2年后硫唑嘌呤或甲氨蝶呤逐渐减量,停用利妥昔单克隆抗体。
总之,皮肤血管炎既可能是单纯性皮肤损害, 也可能是系统性血管炎的皮肤表现。结合特征性肤损害及组织病理表现,诊断不难。辅助检查对于评估内脏受累、潜在疾病以及制定治疗方案和预后判断十分重要。
参考文献:
[1]Carlson JA. The histological assessment of cutaneous vasculitis[J]. Histopathology,2010,56(1):3 ⁃23. DOI:10.1111/j.1365 ⁃2559.2009.03443.x.
[2]Carlson JA,Cavaliere LF,Grant⁃Kels JM. Cutaneous vasculitis:diagnosis and management[J]. Clin Dermatol,2006,24(5):414⁃429. DOI:10.1016/j.clindermatol.2006.07.007.
[3]Shavit E,Alavi A,Sibbald RG. Vasculitis ⁃ what do we have toknow? A review of literature[J]. Int J Low Extrem Wounds,2018,17(4):218⁃226. DOI:10.1177/1534734618804982.
[4]Carlson JA,Ng BT,Chen KR. Cutaneous vasculitis update:diagnostic criteria, classification, epidemiology, etiology,pathogenesis,evaluation and prognosis[J]. Am J Dermatopathol,2005,27(6):504-528. DOI:10.1097 / 01. dad. 0000181109.54532.c5.
[5]Xu LY,Esparza EM,Anadkat MJ,et al. Cutaneous manifestations of vasculitis[J]. Semin Arthritis Rheum,2009,38(5):348⁃360.DOI:10.1016/j.semarthrit.2008.01.007.
[6]Marzano AV,Vezzoli P,Berti E. Skin involvement in cutaneous and systemic vasculitis[J].Autoimmun Rev,2013,12(4):467⁃476. DOI:10.1016/j.autrev.2012.08.005.
[7]Gonzalez⁃Gay MA,Garcia⁃Porrua C,Pujol RM. Clinical approach to cutaneous vasculitis[J]. Curr Opin Rheumatol,2005,17(1):56⁃61. DOI:10.1097/01.bor.0000145519.68725.5a.
[8]廖文俊,樊平申,胡雪慧. 原发性皮肤小血管炎[J].中国麻风皮肤病杂志,2007,23(3):235⁃237. DOI:10.3969/j.issn.1009⁃1157.2007.03.021.Liao WJ,Fan PS,Hu XH. Primary cutaneous small vessel vasculitis[J]. Chin J Lepr Dis,2007,23(3):235 ⁃ 237. DOI:10.3969/j.issn.1009⁃1157.2007.03.021.
[9]Genta MS,Genta RM,Gabay C. Systemic rheumatoid vasculitis:a review[J]. Semin Arthritis Rheum,2006,36(2):88⁃98. DOI:10.1016/j.semarthrit.2006.04.006.
更多参考文献略。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









感谢分享,思路清晰
34