Am J Hematol:MDS的诊断、风险分层和治疗(2023更新)
2023-06-15 聊聊血液 聊聊血液 发表于上海

Myelodysplastic syndromes: 2023 update on diagnosis,risk-stratification, and management”,现整理供各位老师参考。
MDS
骨髓增生异常综合征 (MDS) 包括一组非常异质性的髓系恶性肿瘤,由造血干细胞功能扭曲、炎症和先天性免疫失调、细胞凋亡失调和多种基因组事件引起,分子学改变的组合可导致贫血、感染、出血和转化为急性粒细胞白血病 (AML) 的风险增加。大多数 MDS 患者会死于 MDS 并发症而非转化为AML,因此对 MDS 患者需要独特的治疗策略。更重要的是合并症和克隆性造血与MDS之间的关系,提示 MDS 和发生其他疾病(如心血管疾病)之间存在相互作用。
原始 IPSS 和改良 IPSS-R 是最常用的分层系统,纳入分子学数据的 IPSS-M也已出现并进行验证。在对 MDS 患者做出治疗决策时,还需要其他几个重要因素,包括患者年龄、合并症的类型和严重程度、血细胞减少的意义和数量、输血需求、特定基因组改变的存在(现在通过 IPSS-M 计算)、原始细胞百分比、细胞遗传学特征、异基因干细胞移植 (alloSCT) 的可能性,尤其重要的是去甲基化药物 (HMA) 治疗史,因为接受 HMA 治疗的 MDS 患者的生物学和自然史与既往未接受过HMA治疗的患者存在很大差异,即使IPSS 和 IPSS-R 评分相似。
近日《American Journal of Hematology》发表了MD安德森癌症中心Guillermo Garcia-Manero教授的综述“Myelodysplastic syndromes: 2023 update on diagnosis,risk-stratification, and management”,现整理供各位老师参考,水平有限敬请谅解。

诊断
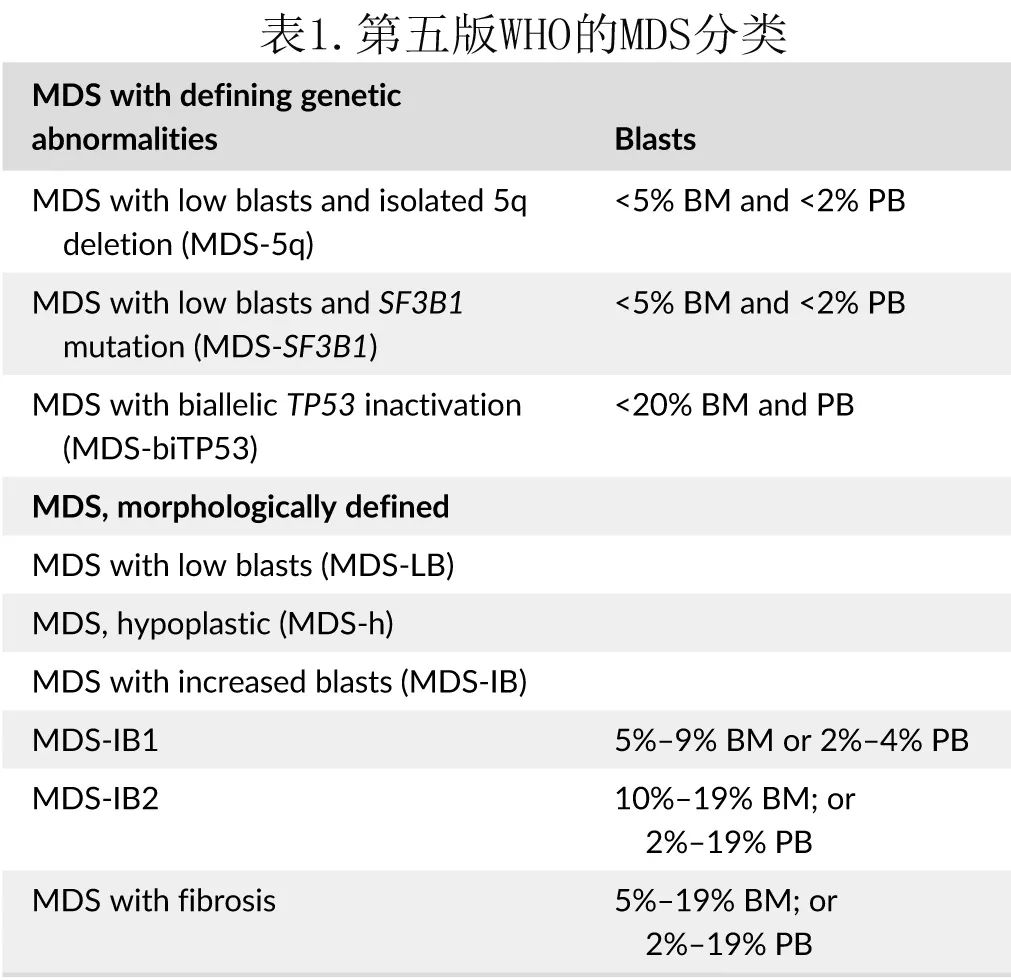
一般由血细胞减少怀疑 MDS,并通过骨髓穿刺和活检确诊,骨髓穿刺可详细评估细胞形态和原始细胞百分比,骨髓活检可用于测定骨髓细胞构成和结构,通过存在异型增生可确诊。MDS有许多形态学分类,最近的是2022年 WHO 分类(表1),根据该分类MDS现在称为骨髓增生异常肿瘤(myelodysplastic neoplasm),但首字母缩写仍为MDS。作者认为改变名字是一个错误,因为这些疾病显然是综合征,而不仅仅是肿瘤,包括其与合并症的相关性便可证明这一点。
此外ICC也提出了类似的理论。作者认为MDS 中原始细胞的百分比仍应考虑<20%,只因MDS与 AML 患者的预后相似而将原始细胞百分比降低至10%是没有科学意义的,因为预后不等同于诊断。假如按照上述理论,所有具有高危异常(即 p53 突变)的疾病便可以使用同一个名字来描述(包括实体瘤和血液肿瘤),仅仅是因为它们预后较差,或可以靶向它(即 AML 和胶质母细胞瘤中的 IDH 突变)。此外,形态学上 MDS 也不同于AML。
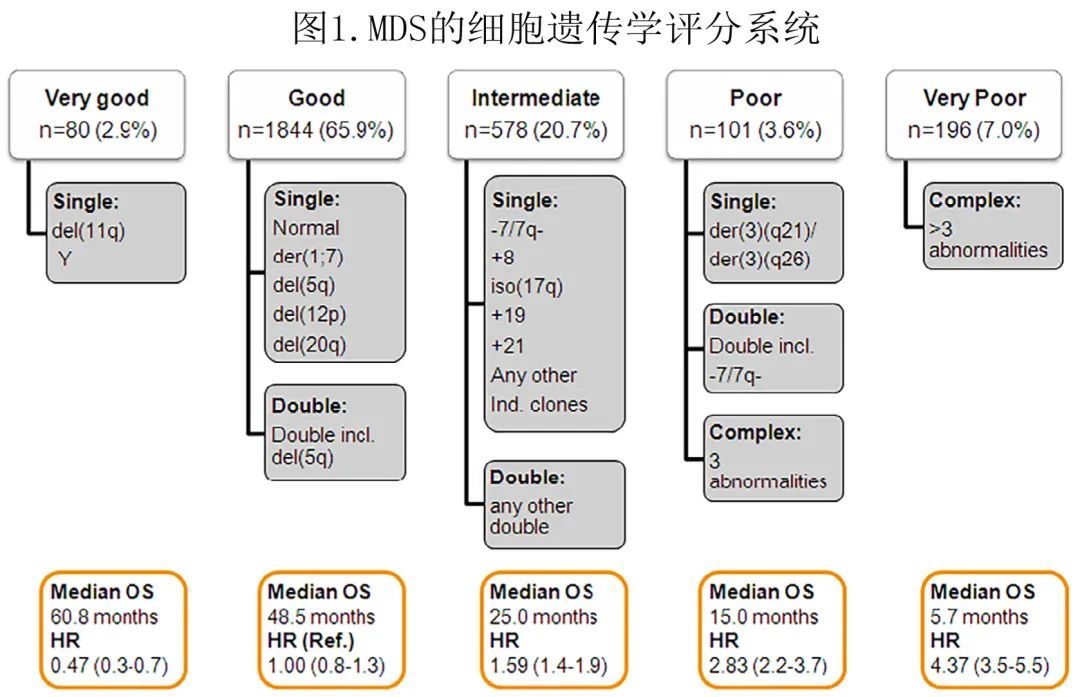
完成 MDS 患者的实验室评估还需要进行多项额外检查。细胞遗传学模式在 MDS 中具有非常高的异质性,细胞遗传学对于计算患者预后和在部分患者亚组中选择最有效治疗具有重要意义。MDS 的最新细胞遗传学风险分类包括5个不同的亚组,包括20种不同的基因异常(图1)。但MDS 的细胞遗传学模式不稳定,相当一部分患者可随着疾病进展获得额外的细胞遗传学异常,这一现象与向 AML 转化的风险增加和较差的生存期相关。据报道,评估染色体大结构变化的新技术,如光学基因组图谱,能够进行细胞遗传学分析而不需要培养骨髓细胞或条带解释,从而降低检测失败的比例。
许多其他检测方法可辅助诊断MDS,包括流式细胞术、荧光原位杂交 (FISH) 和基因组测序技术。流式细胞术可以识别异常表型模式,并有助于微小异型增生患者。由于 MDS 细胞遗传学改变的显著异质性,没有证据表明一组 FISH 探针可以替代常规的20分裂象(metaphase)细胞遗传学分析。因此作者认为,FISH 和流式细胞术不应视为 MDS 患者标准检查程序的一部分,应在特定情况下使用。得益于 MDS 中多种基因突变的发现,现在可以获得 MDS 患者分子注释(molecular annotation)的临床工具,可用于补充其他诊断工具。大多数 MDS 患者携带 DNA 甲基化、染色质调控、RNA剪接、转录调控、DNA修复、黏连蛋白功能或信号转导相关基因的一个或多个体细胞突变。
ICUS、潜质未定的克隆性造血和CCUS
由于 MDS 的诊断是基于形态学评估,它可能是主观的,特别是在早期低危疾病和轻微异型增生的患者中。近20%的患者在初次就诊时可发生诊断差异,这对治疗决策和患者咨询具有明显的意义。一般而言,原始细胞过多的患者诊断很明确;而问题在于没有过多原始细胞的患者中,其诊断是基于异型增生。需要对异型增生证据极少或没有异型增生诊断证据的患者进行临床评估,这些情况下建议排除血细胞减少的其他诱因,常规检查应包括分析贫血和血小板减少症,并排除失血或炎症过程。怀疑MDS时需要考虑胃肠道检查。一旦排除了血细胞减少的其他潜在诱因,额外的诊断工具包括细胞遗传学评估、流式细胞术和DNA 测序,可以帮助定义诊断和预测患者结局。血细胞减少但无异型增生的患者是意义未明的特发性血细胞减少症(ICUS),其中部分患者可能存在细胞遗传学异常或髓系恶性肿瘤常见的体细胞突变。在无 MDS 诊断标准但血细胞减少的背景下存在体细胞突变,目前认为是意义未明的克隆性血细胞减少症(CCUS);这一区别得到了以下数据的支持:尽管接近25%的 ICUS 患者最终可能发展为 MDS 或AML,但在存在克隆突变的情况下该风险5年时可从9%显著增加至82%,尤其是存在高度预测性突变模式的情况下。
因此,ICUS、CCUS和 MDS 之间的详细评估和仔细鉴别诊断至关重要(表2)。在无血液学疾病证据的老年个体造血细胞中也报告了克隆性体细胞突变,属于与年龄相关的潜质未定的克隆性造血(CHIP,clonal hematopoiesis of indetermined potential)。克隆性造血也是治疗相关 MDS 和 有AML 的风险因素,有克隆性造血的患者发生 MDS 的风险也增加。对 CHIP 或 CCUS 患者的评估和潜在治疗很有意义,这来自其他全身性疾病(例如心血管疾病)和 CHIP/CCUS 之间的关系,以及通过治疗 CHIP/CCUS 可以预防进展为髓系肿瘤 (MN) 的假设。许多中心正在开发“CHIP诊所”和临床试验。潜在的靶点包括IL-1、剪接改变和 IDH 突变等。最近,Weeks等提出了 CHIP/CCUS 的预后模型。此外,ASH 2022上提供的数据表明,CHIP/CCUS中最常见的突变是DNMT3A,与进展为 MN 的频率相对较低相关;该数据可能引领正确选择需要治疗或更强化的干预的CHIP/CCUS 患者。

一类重要的患者是具有 MDS/MPN 特征的患者,这些患者有骨髓增生性成分的证据(伴或不伴纤维化)。目前除慢性粒单核细胞白血病外尚未完全了解 MDS/MPN 患者的自然史,作者目前将其按 MDS 治疗,但研究正在进行中以明确这一问题。值得注意的是,特定 MDS/MPN 亚型具有特殊的突变特征,可能获益于特定治疗,例如,伴环形铁粒幼细胞和血小板增多的 MDS/MPN 患者通常存在 SF3B1 和 JAK2 突变,对来那度胺治疗可能有良好反应。
风险分层
MDS 患者的预后具有很大的异质性,因此需要开发预后系统以进行风险分层,并有助于治疗的时机和选择;除了形态学分类的内在预后作用外,还有许多预后评分正在用于MDS。1997年后一直实行IPSS的已基本被 IPSS-R 取代,与 IPSS 相比,IPSS-R包括不同的血细胞减少临界点,并结合了更全面的细胞遗传学评分,IPSS-R 现在是评估风险的标准工具。IPSS-M 最近发表并得到了许多中心的验证,包括关于p53(单等位基因与双等位基因突变)的详细信息并纳入16个基因;但该工具尚未在特定患者亚组中进行测试,例如接受去甲基化药物治疗或 HMA 治疗失败的患者。
低危病患者的自然史具有非常大的异质性,作者通过 IPSS 评估大样本低危或中危-1 患者的结局,发现低危 MDS 患者的预后差异显著,并开发了低危 MDS 的特异性预后评分,对于开发针对低危患者的特定干预措施具有重大影响。该模型已在多个场合得到验证,并用于识别预后不良的低危患者,这些患者可能是早期干预的候选者。Bejar等认为,预后较差和疾病风险较低的患者会比风险较好的患者积累更多的突变事件,该数据为识别该组患者和较差预后提供了潜在的分子学基础。新的 IPSS-M 模型应该真正有助于明确伴低危特征(如二倍体细胞遗传学和原始细胞百分比低)患者的预后。
MDS 好发于患有合并症的老年患者,而上文讨论的预后系统在计算 MDS 患者的自然史时均未包括合并症的影响。为研究该问题,作者在500例 MDS 患者队列中使用了一种称为 ACE-27 的综合合并症评分。合并症对生存率有显著的独立影响,因此可以开发一个包括年龄、IPSS和 ACE-27 评分的预后评分,而研究中使用 IPSS-R 联合ACE-27也可获得相同的结果,表明MDS需要增加合并症评分。
其他预后评分系统包括增生低下型MDS 和治疗相关 MDS的评分系统,但传统风险分层模型(如IPSS-R、MDACC模型或WPSS)在治疗相关MDS中似乎同样有效。需要注意的是,t-MDS的预后与细胞遗传学异常密切相关:二倍体患者的预后与初治患者无差异。
细胞遗传学和分子学异常
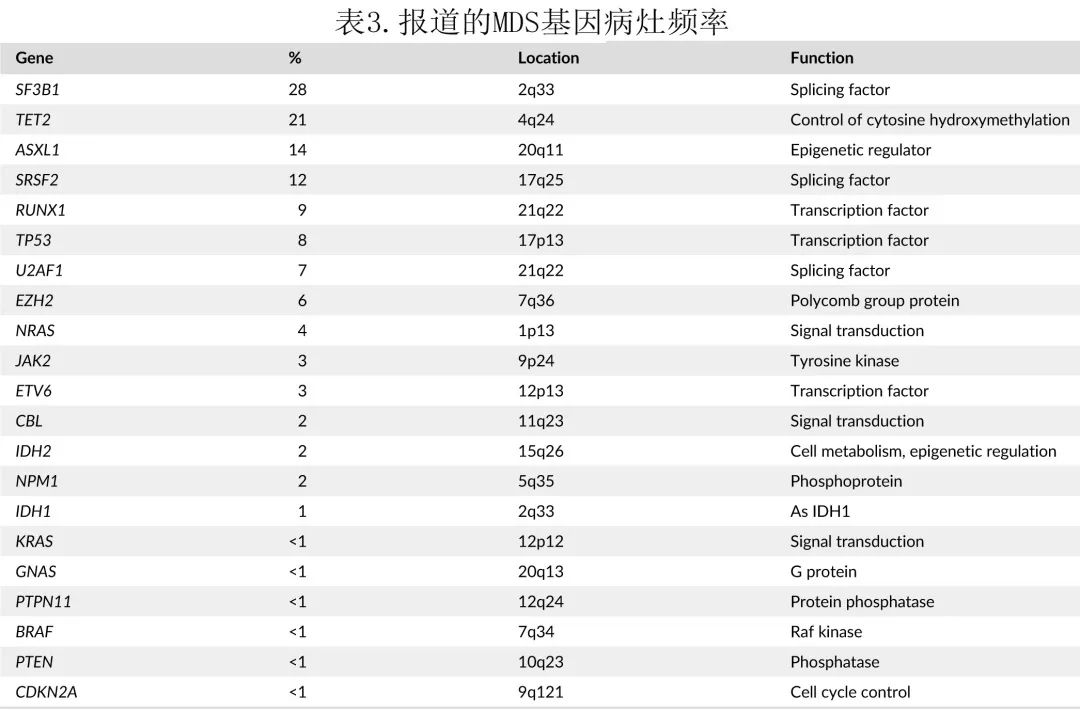
常见突变频率见表3。尽管研究存在异质性,但RUNX1、TP53或 EZH 等基因突变始终与不良预后相关,而剪接因子 SF3B1 突变与有利结局和延长生存期相关。
风险分层治疗
2023年MDS治疗的当前概念框架
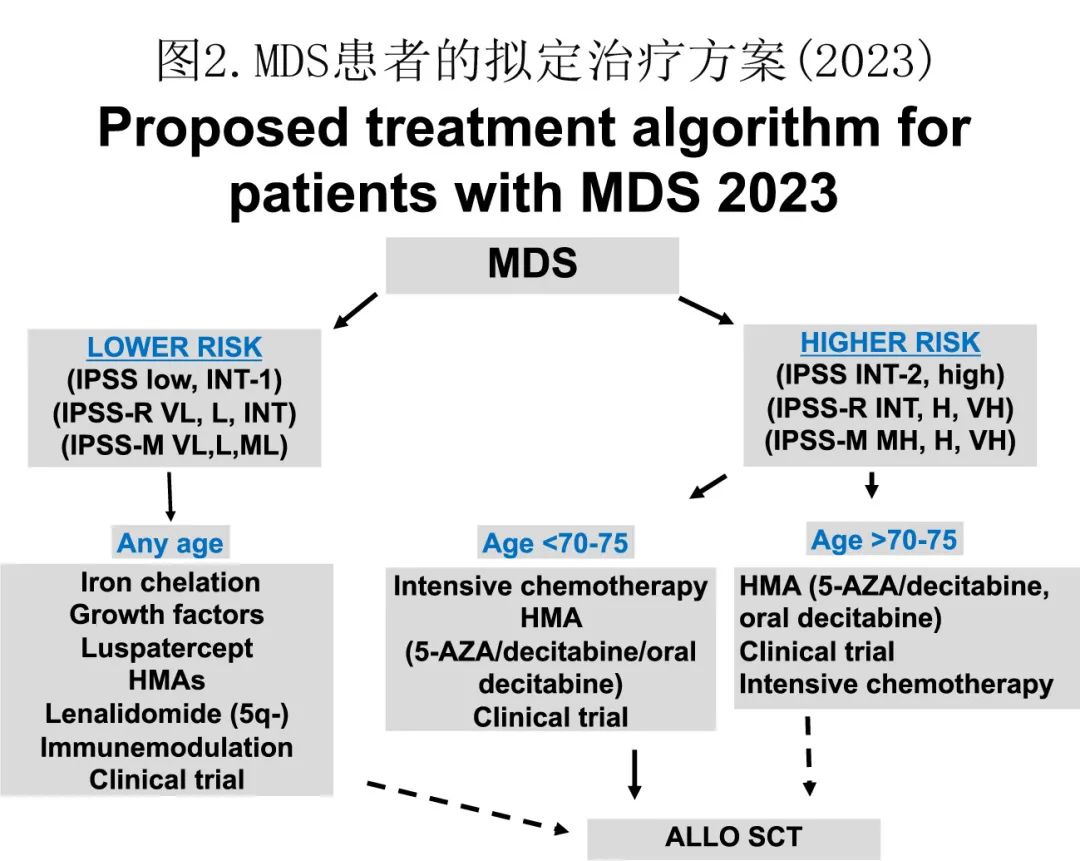
作者将患者分为6类。首先是无 MDS 形态学诊断的患者亚组,包括ICUS、CHIP和CCUS。接下来将 MDS 患者分为低危或高危,但根据其是否曾暴露于 HMA 进一步细分:低危MDS、低危 HMA 治疗失败、高危和高危 HMA 治疗失败。最后一组预后极差的患者是由 MDS 演变而来的AML患者,尤其是在基于 HMA 的治疗后;但本综述中不讨论最后一个患者组。此外,临床分级基因组有助于所有这些患者亚组的治疗决策。下文进一步讨论这5个亚组患者。图2总结了 MDS 患者的当前治疗策略。低危患者的治疗目标与高危患者和 HMA 治疗失败患者不同:低危患者目标是减少输血需求和向高危疾病或 AML 的转化,以及改善生存率;高危患者目标是延长生存。
ICUS/CHIP/CCUS的治疗
建议:ICUS/CHIP/CCUS患者不应接受治疗,而应优先在专门的“CHIP”诊所接受随访。应注意合并症的护理。如适用,可考虑进行临床试验
低危MDS的治疗
该亚组患者的治疗是基于输血需求。一般而言,通常对非输血依赖性患者进行观察,直至输血依赖,但这一概念目前正在受到挑战。以下是目前可用于低危 MDS 患者的药物建议总结。
建议:
红细胞生长因子支持:在大多数伴显著贫血而无其他血细胞减少的低危 MDS 患者中,一系列红细胞生成刺激剂(ESA)加或不加 G-CSF并非禁忌,数据表明早期加入这些药物更有效。作者会维持治疗至少3个月以判断疗效,在有反应的患者中继续治疗,直至失去输注效果。
罗特西普:对ESA无应答、失去应答或不适合ESA的 RARS 患者的标准治疗。
来那度胺:来那度胺在低危MDS、贫血、良好血小板和 del5q 患者中的缓解程度使其成为该患者亚组的标准治疗,缓解患者的生存期数据进一步证实了这一点。作者不考虑将该药物用于血小板减少症患者。基于 III 期随机研究的结果,来那度胺可认为是特定非 del5q MDS 红细胞输注依赖性患者的一种选择。建议在开始来那度胺治疗前评估 del5q MDS 患者的 TP53 突变状态。尽管目前的证据不支持 TP53 突变患者暂停来那度胺治疗,但应密切监测这些患者的疗效不足、无效或进展体征。Sintra-Rev 研究的最终报告也支持在 del5q-MDS 非输血依赖患者中使用低剂量来那度胺。
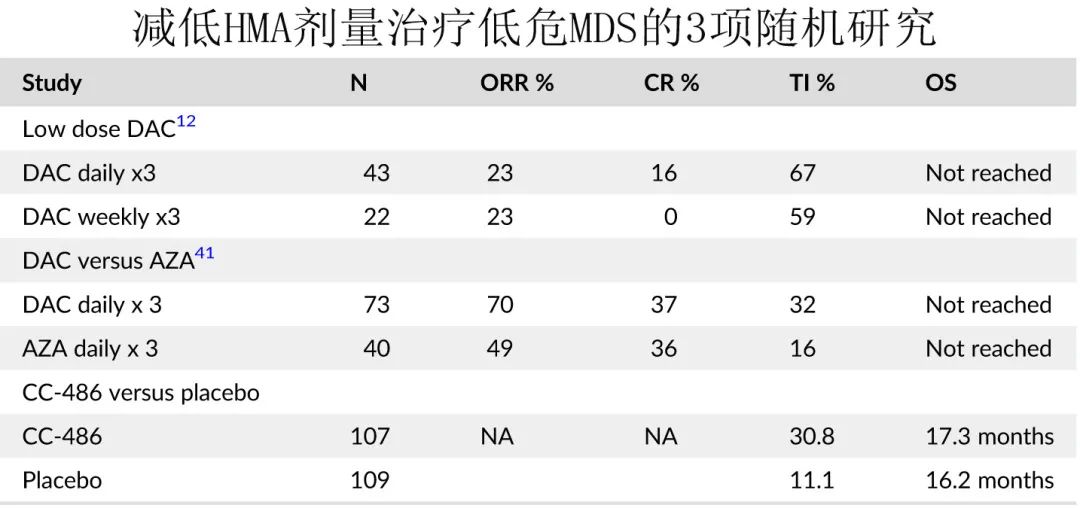
氮杂核苷类:阿扎胞苷和地西他滨均在美国用于输血依赖性低危患者,大多数接受这些药物治疗的患者为生长因子支持或来那度胺治疗失败或不适合的患者。尽管尚需要随机研究的结果来确定这些药物是否可以改变低危疾病的自然史,并作为一线治疗或生长因子支持后的标准治疗,但最近的数据支持低剂量使用,尤其是在具有更多不良特征的患者中。未来口服 HMA 可能在低危 MDS 中发挥作用。
免疫治疗:大多数老年患者无法耐受抗胸腺细胞球蛋白,多数患者接受某种形式的支持治疗,包括环孢素、生长因子和类固醇,但其影响也未知。年轻的重度发育不良 MDS 患者应尽快考虑alloSCT;对于不适合移植的患者,建议联合使用马抗胸腺细胞球蛋白。在报告其他临床试验的更多数据之前,目前不推荐使用阿仑单抗。此外艾曲波帕的数据值得关注。
AlloSCT:通常不建议初诊时风险较低患者进行alloSCT,也就是说,由于供者鉴定所需的时间,作者将所有潜在的候选患者转诊进行移植咨询,以预测未来的需求。对于适合 alloSCT 且接受过多种治疗(生长因子、来那度胺、阿扎胞苷等)的患者,应考虑进行移植。这些患者也是临床试验的候选者。年轻的发育不良 MDS 患者应考虑提前进行alloSCT。
MDS的支持性治疗措施:对于未接受细胞毒或免疫抑制治疗的孤立性中性粒细胞减少症和 MDS 患者,作者不常规推荐使用抗生素;对接受积极治疗的患者使用预防性抗生素。在铁蛋白水平超过 2500 ng/mL 的患者中使用铁螯合,但会考虑这些患者参加铁螯合的临床试验。
促血小板生成素
建议:应谨慎使用促血小板生成素,可能仅用于无其他选择的难治性患者
复发或难治性低危MDS的治疗选择以及低危MDS的试验性新药
建议:目前尚无药物获批用于治疗 HMA 治疗失败的低危 MDS 患者,唯一的有效选项是alloSCT。否则应考虑患者参加试验性临床试验。
新诊断高危MDS的治疗选择
建议:
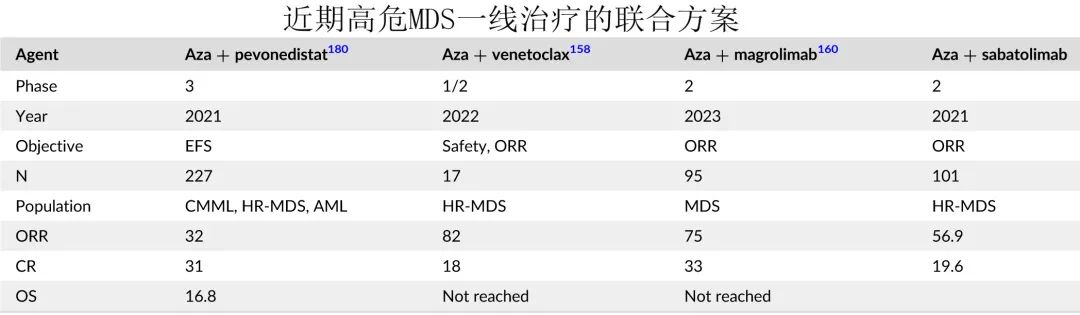
氮杂核苷类:氮杂核苷类是大多数高危患者的标准治疗。尚无研究比较阿扎胞苷与地西他滨,尽管缓解率似乎相似,但在一项随机试验中,仅阿扎胞苷可改善生存期。基于此,考虑阿扎胞苷标准治疗作为高危 MDS 的一线治疗。
AML样化疗:随机 AZA-001 研究没有足够效力(AML样治疗的样本量过小)证明阿扎胞苷相对于 AML 样治疗的优效性,其原因是大多数研究者不认为其患者适合AML样治疗,因此问题在于谁可能适合AML 治疗。在作者的实践中,仅限于对治疗反应可能性较高的年轻患者,如二倍体患者或 alloSCT 候选者。作者很少在老年患者或细胞遗传学高危或 TP53 突变患者中使用 AML 治疗。
AlloSCT:应告知所有可能接受 alloSCT 的患者移植的可能性,最好是患者入组 alloSCT 的 MDS临床试验。尽管高危 MDS 患者应考虑alloSCT,但在高危突变(如TP53)患者中可能并非如此,对于这类患者,作者认为只有最佳供者可用、获得最佳移植前缓解、以及考虑低剂量移植后阿扎胞苷或地西他滨时,才可能考虑移植。移植后复发高危患者也可考虑使用低剂量阿扎胞苷。作者赞同最近提出的美国指南(指南参见DeFilipp Z, Ciurea SO, Cutler C, et al. Hematopoietic cell transplantation in the management of Myelodysplastic syndrome: an evidence-based review from the American Society for Transplantation and Cellular Therapy Committee on practice guidelines. Transplant Cell Ther. 2023;29(2):71-81.)。
高危MDS患者的试验性方案
建议:所有 MDS 患者应尽可能考虑参加试验性临床试验。
HMA治疗失败仍是MDS的主要未满足需求
建议:HMA治疗失败MDS的预后极差,目前尚无疗法显示对该组患者有显著疗效。所有复发或难治性的高危患者均应考虑参加试验性临床试验和alloSCT。
将精准医学纳入MDS
建议:提倡在基线和每次对 MDS 患者做出治疗决策时使用NGS panel。
参考文献
Garcia-Manero G.Myelodysplastic syndromes: update on diagnosis, risk-stratification, and management.Am J Hematol . 2023 Jun 8. doi: 10.1002/ajh.26984.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言