酪氨酸血症Ⅰ型:症状与体征、病因、流行病学、诊断和治疗
2022-08-27 MedSci原创 MedSci原创

酪氨酸血症(tyrosinemia)是一种罕见的常染色体隐性遗传代谢病,由于酪氨酸降解障碍导致脑、肝、肾、骨骼等多脏器损害,预后不良,致死及致残率很高。不同类型的患者临床表现不同,低酪氨酸饮食、药物治
酪氨酸血症(tyrosinemia)是一种罕见的常染色体隐性遗传代谢病,由于酪氨酸降解障碍导致脑、肝、肾、骨骼等多脏器损害,预后不良,致死及致残率很高。不同类型的患者临床表现不同,低酪氨酸饮食、药物治疗是主要的干预方法,必要时需通过肝移植治疗。
根据缺陷酶的不同,酪氨酸血症分为三型:
1.酪氨酸血症Ⅰ型是由于FAH基因突变导致酪氨酸代谢过程的终末酶延胡索酰乙酰乙酸水解酶缺陷,酪氨酸及其代谢产物琥珀酰丙酮、4-羟基苯乳酸及4-羟基苯丙酮酸等蓄积。
2.酪氨酸血症Ⅱ型是由于酪氨酸转氨酶缺乏所致酪氨酸分解障碍。
3.酪氨酸血症Ⅲ型是由于4-羟基苯丙酮酸二氧化酶缺乏所致疾病。
酪氨酸血症Ⅰ型,也被称为肝肾酪氨酸血症(hepatorenal tyrosinemia,HT-1),为延胡索酰乙酰乙酸水解酶(fumarylacetoacetate hydrolase,FAH) 缺陷所致,以肝、肾和周围神经病变为特征。酪氨酸血症Ⅱ型,为酪氨酸氨基转移酶(tyrosine aminotransferase,TAT) 缺陷所致,以角膜增厚、掌跖角化和发育落后为特征。酪氨酸血症Ⅲ型,极为罕见,为4- 羟基苯丙酮酸双加氧酶(hydroxyphenylpyruvic acid dioxygenase,HPPD) 缺陷所致,以神经精神症状为主要表现。鉴于酪氨酸血症Ⅰ型病例报道相对较多,且对健康危害较大,本文将主要介绍酪氨酸血症Ⅰ型。
一、一般概述
I 型酪氨酸血症是一种罕见的常染色体隐性遗传代谢疾病,其特征是缺乏最终分解氨基酸酪氨酸所需的富马酰乙酰乙酸水解酶 (FAH)。未能正确分解酪氨酸会导致酪氨酸及其代谢物在肝脏中异常积聚,从而可能导致严重的肝脏疾病。酪氨酸也可能积聚在肾脏和中枢神经系统中。
与 I 型酪氨酸血症相关的症状和体格检查结果出现在出生后的头几个月,包括体重增加和未按预期速度生长(发育迟缓)、发烧、腹泻、呕吐、肝脏异常肿大(肝肿大)和发黄皮肤和眼白(黄疸)。如果不及时治疗,I 型酪氨酸血症可能会发展为更严重的并发症,例如严重的肝病、肝硬化和肝癌。确诊后应尽快开始使用尼替西农和低酪氨酸饮食进行治疗。
二、症状与体征
与 I 型酪氨酸血症相关的症状通常因人而异。患有 I 型酪氨酸血症的婴儿通常表现为急性或慢性疾病。
I 型酪氨酸血症的急性形式在出生时(先天性)或生命的最初几个月就存在。这种形式的疾病比慢性形式更常见和更严重。急性形式的婴儿表现出快速发作的症状,通常始于体重增加和以预期速度生长(未能茁壮成长)。其他早期症状包括发烧、腹泻、血便(黑便)和呕吐。受影响的婴儿还可能表现出肝脏异常扩大(肝肿大)、容易瘀伤、黄疸、嗜睡和/或易怒。一些受影响的婴儿可能会产生独特的卷心菜气味。
最终,患有 I 型急性酪氨酸血症的婴儿会出现发育迟缓、脾脏异常肿大(脾肿大)和腹部积液(水肿)(腹水)。该疾病可能迅速发展为急性危及生命的肝功能衰竭和凝血异常(凝血障碍)。
I型酪氨酸血症的慢性形式比急性形式的发生频率低,其特征是起病更缓慢,症状表现更不严重。 I 型酪氨酸血症的症状可能在患有慢性疾病的婴儿中直到六个月后才会变得明显。未能茁壮成长通常是第一个症状。其他症状包括发育迟缓和进行性瘢痕形成和肝脏功能受损(肝硬化),导致慢性肝功能衰竭。
许多患有 I 型酪氨酸血症的婴儿会出现肾脏(肾脏)异常,例如肾范可尼综合征,这是一种以肾功能不全为特征的罕见疾病,通常会导致骨骼结构进行性软化和弱化(佝偻病)。 Fanconi 综合征也与呕吐、脱水、虚弱和发烧有关。
大约 40% 的受影响婴儿也会在轻微感染后出现影响许多神经的疾病(多发性神经病)。这些发作可能被称为神经系统危机,与腿部和胃部剧烈疼痛、肌肉张力增加(高张力)、呕吐、肠梗阻(肠梗阻)、心律不齐(心动过速)和高血压力(高血压)。在这些发作期间,一些受影响的个体也可能表现出自残行为(例如,咬舌头或磨牙)。可能发生神经危象和呼吸衰竭。
受影响的婴儿还可能出现分隔心脏左心室和右心室的隔板增大(肥大),在某些儿童中,左心室壁可能会增大(肥厚性心肌病)。此外,受影响的婴儿和儿童比普通人群更容易患上一种称为肝细胞癌的肝癌。
用尼替西农和低酪氨酸饮食治疗受影响的儿童已将存活率提高到 90% 以上,并导致正常生长、改善肝功能、预防肝硬化、纠正肾脏疾病和改善佝偻病。
三、病因
酪氨酸血症是由负责产生 FAH 酶的延胡索乙酰乙酸水解酶 (FAH) 基因突变引起的。这种酶的缺乏会导致富马酰乙酰乙酸的积累和酪氨酸及其代谢物在肝脏、肾脏和中枢神经系统中的积累,最终导致 I 型酪氨酸血症。
I型酪氨酸血症作为常染色体隐性遗传病遗传。
隐性遗传疾病发生在个体遗传了同一性状的异常基因的两个拷贝时,每个拷贝来自父母一方。如果一个人接受一个正常基因和一个疾病基因,该人将成为该疾病的携带者,但通常不会出现症状。每次怀孕,两个携带者父母都通过缺陷基因并有受影响的孩子的风险是 25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是 50%。一个孩子从父母双方那里获得正常基因并在该特定特征上遗传正常的机会是 25%。男性和女性的风险相同。
近亲(近亲)的父母比无关父母携带相同异常基因的可能性更高,这增加了患隐性遗传疾病的孩子的风险。
四、流行病学
I 型酪氨酸血症影响男性和女性的数量相等。 酪氨酸血症Ⅰ型为常染色体隐性遗传病,发病率为 1/120 000~1/100 000。美国人群的突变携带频率 1/150~1/100,由于奠基者效应,斯堪的纳维亚半岛酪氨酸血症Ⅰ型的活产新生儿发病率约为 1/74 000,芬兰和挪威约为 1/60 000。此外,在加拿大的魁北克省,活产新生儿发病率约为 1/16 000,携带频率约为 1/66。 据估计,魁北克省 Saguenay-Lac Saint-Jean 地区的新生儿患病率为 1,850 分之一。中国尚缺少相关流行病学资料。
五、鉴别诊断
以下疾病的体征和症状可能与 I 型酪氨酸血症相似。比较可能有助于鉴别诊断:
急性间歇性卟啉症 (AIP) 是一组称为卟啉症的遗传性代谢疾病之一。 AIP 的特点是缺乏胆色素原脱氨酶 (PBG-D),也称为尿卟啉原 I-合酶。症状可能包括严重的腹痛、恶心、呕吐和便秘。也可能出现类似于 I 型酪氨酸血症的神经系统症状,包括手臂和腿部疼痛、肌肉无力、心率加快和血压升高。受影响的人也可能会出现幻觉和癫痫发作。 AIP 是一种常染色体隐性遗传疾病
半乳糖血症是一种罕见的遗传性碳水化合物代谢疾病,会影响身体将半乳糖(牛奶中包含的一种糖,包括人类母乳)转化为葡萄糖(一种不同类型的糖)的能力。半乳糖通过一系列三种酶反应转化为葡萄糖。半乳糖血症是由于缺乏对这一过程至关重要的 1-磷酸半乳糖尿苷转移酶所致。患有半乳糖血症的婴儿在出生时看起来很正常,但在几天或几周内就会失去食欲(厌食)并开始过度呕吐。皮肤和眼白发黄(黄疸)、肝脏肿大(肝肿大)、体重增加和以预期速度生长(发育迟缓),最终导致腹腔积液(腹水)和其他肿胀(水肿)也可能发生。半乳糖血症作为常染色体隐性遗传病遗传。
遗传性果糖不耐症 (HFI) 是一种罕见的遗传性无法消化果糖(水果糖)或其前体(糖、山梨糖醇和红糖)的疾病。这种疾病是由于缺乏或缺乏 1-磷酸果糖醛缩酶,导致肝脏、肾脏和小肠中 1-磷酸果糖的积累。患有 HFI 的婴儿通常会对糖果和水果产生强烈的反感。在 HFI 婴儿的饮食中添加果糖后不久,症状通常会变得明显。这些可能包括长时间呕吐、无法茁壮成长以及皮肤和眼白发黄(黄疸)。其他症状包括肝脏肿大(肝肿大)和由于身体形成血栓的能力异常而导致胃肠道出血的趋势。
酪氨酸血症 II 型是由酪氨酸氨基转移酶缺乏引起的常染色体隐性遗传疾病。受影响的儿童没有肝脏异常。症状可能包括眼睛过度流泪、眼睛疼痛和发红,以及手掌和脚底疼痛的皮肤损伤和对光敏感(畏光)。有时会出现发育迟缓和智力障碍。低苯丙氨酸和酪氨酸饮食可改善症状。
III 型酪氨酸血症是一种常染色体隐性遗传疾病,由 4-羟苯基丙酮酸双加氧酶缺乏引起。受影响的儿童没有肝脏异常。这种情况极为罕见,很少有儿童被描述,但症状可能包括失去平衡和协调(共济失调)、皮肤和眼睛异常以及发育迟缓。低苯丙氨酸和酪氨酸饮食可改善症状。
六、诊断
I 型酪氨酸血症的诊断基于全面的临床评估、详细的患者病史和专门的测试。如果婴儿在出生后头三个月表现出发育迟缓和肝脏肿大(肝肿大),则可能怀疑 I 型酪氨酸血症的诊断。当在尿液中检测到酪氨酸代谢物和琥珀酰丙酮时,很可能进行诊断。也可以根据肝组织或培养的成纤维细胞中 FAH 活性的降低来做出诊断,但这种测试并不容易获得。 FAH 基因突变的分子遗传学检测可用于确诊。
I 型酪氨酸血症也可以通过新生儿筛查项目进行诊断。琥珀酰丙酮可以通过串联质谱在新生儿血斑上进行测量。美国的大多数州对每个新生儿进行 1 型酪氨酸血症筛查。早期发现很重要,因为及时发现和治疗可以防止婴儿期出现严重问题。
如果已在家庭中鉴定出特定的基因突变,则可以通过 DNA 分析进行携带者检测和产前诊断。下一代 DNA 测序技术,如外显子组测序、全基因组测序 (WGS) 可以帮助识别导致疾病的突变。通过检测羊水中的琥珀酰丙酮和 DNA 分析也可以进行产前诊断。
诊疗流程
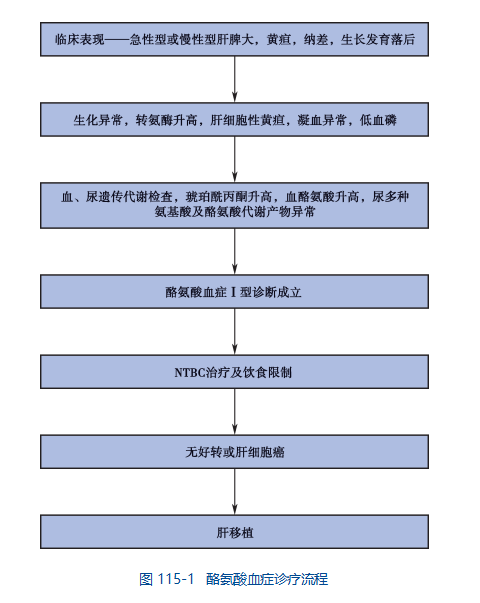
七、治疗
2017年,Nityr(尼替西农片剂)被美国食品药品监督管理局(FDA)批准用于治疗1型遗传性酪氨酸血症。Nityr由Cycle Pharmaceuticals生产。
2002 年,FDA 批准了孤儿药 Orfadin,一种尼替西农的胶囊和口服混悬剂,用于治疗 I 型酪氨酸血症。尼替西农由瑞典孤儿国际 Biovitrum AB 开发,由 Sobi, Inc. 上市。
这些药物只能由有治疗 I 型酪氨酸血症经验的医生开具处方,因为必须根据特定的生化测试和体重为每位患者调整正确的剂量。获得擅长管理需要低蛋白饮食的先天性新陈代谢错误儿童的营养师是治疗的重要组成部分。应定期监测血液检查以维持患者的正确剂量。
尼替西农必须与限制氨基酸酪氨酸和苯丙氨酸的饮食一起使用。确诊后应尽快开始尼替西农治疗和饮食管理。
患有 I 型酪氨酸血症的婴儿被置于含有有限量苯丙氨酸和酪氨酸的低蛋白饮食中。一些受影响的婴儿仅通过饮食管理就表现出肝脏和肾脏异常的改善。然而,仍有可能进展为肝硬化、肝功能衰竭和潜在的肝细胞癌。医生经常建议受影响的人在其一生中使用特殊的医疗食品严格饮食。
对于在诊断时已经发展为终末期肝功能衰竭、有肝癌(肝细胞癌)证据或对尼替西农治疗无反应的受影响婴儿,可能需要进行肝移植。在一些儿童中,肝移植可改善肾功能。
建议对受影响的个人及其家人进行遗传咨询。其他治疗是对症和支持性的。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
https://www.chard.org.cn/#/knowledge/jbzsk/detail/149
Masurel-Paulet A, Poggi-Bach J, Rolland MO, et al. NTBC treatment in tyrosinaemia type 1: long-term outcome in French patients. J Inherit Metab Dis. 2008; 31: 81-7.
McKiernan PKJ. Nitisinone in the treatment of hereditary tyrosinaemia type 1. Drugs. 2006; 66: 743-50.
Rashed MS, Al-Ahaidib LY, Al-Dirbashi OY, et al. Tandem mass spectrometric assay of succinylacetone in urine for the diagnosis of hepatorenal tyrosinemia. Anal Biochem. 2005; 339: 310-7.
Pierik LJ, van Spronsen FJ, Bijleveld CM, van Dael CM. Renal function in tyrosinaemia type I after liver transplantation: a long-term follow-up. J Inherit Metab Dis. 2005; 28: 871-6.
van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifl). J Pediatr Gastroenterol Nutr. 2005; 40: 90-3.
Russo P, Mitchell GA, Tanguay RM. Tyrosinemia: a review. Pediatr Dev Pathol 2001;4: 212-21.
Barkaoui E, Debray D, Habès D, Ogier H, Bernard O.Favorable outcome of treatment of NTBC of acute liver insufficiency disclosing hereditary tyrosinemia type I. Arch Pediatr. 1999;6:540-44.
Croffie JM, Gupta SK, Chong SK, Fitzgerald JF. Tyrosinemia type I should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure. Pediatrics. 1999;103:675-8.
Forget S, Patriquin HB, Dubois J, et al. The kidney in children with tyrosinemia: sonographic, CT and biochemical findings. Pediatr Radiol. 1999;29:104-08.
Burton BK. Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics. 1998;102:E69.
Poudrier J, Lettre F, Scriver CR, Larochelle J, Tanguay RM. Different clinical forms of hereditary tyrosinemia (type I) in patients with identical genotypes. Mol Genet Metab. 1998;64:119-25.
St-Louis M, Tanguay RM. Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: overview. Hum Mutat. 1997;9:291-99.
Paradis K Tyrosinemia: the Quebec experience. Clin Invest Med. 1996;19:311-16.
Holme E, Lindstedt S. Diagnosis and management of tyrosinemia type I. Curr Opin Pediatr. 1995;7:726-32.
van Spronsen FJ, Thomasse Y, Smit GP et al. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187-90.
Sniderman King L, Trahms C, Scott CR. Tyrosinemia Type I. 2006 Jul 24 [Updated 2017 May 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1515/ Accessed Sept 10, 2019.
McKusick VA, ed. Online Mendelian Inheritance in Man (OMIM). Baltimore, MD: The Johns Hopkins University; Entry No. 276700; Last Update: 06/12/2019. https://www.omim.org/entry/276700 Accessed 9/10/19.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#流行病#
30