Aarskog-Scott综合征:症状体征、病因、流行病学、诊断和治疗
2022-08-31 MedSci原创 MedSci原创

Aarskog-Scott综合征也称为Aarskog综合征、阿尔珀斯氏综合征、阿-斯氏综合征、面指生殖器综合征、或面源性发育不良是一种罕见的X连锁疾病,主要影响男性,其特征是面部、骨骼和生殖器异常。
Aarskog-Scott综合征也称为Aarskog综合征、阿尔珀斯氏综合征、阿-斯氏综合征、面指生殖器综合征、或面源性发育不良是一种罕见的X连锁疾病,主要影响男性,其特征是面部、骨骼和生殖器异常。
1970年,挪威的Aarskog描述了一种综合征,在同一家族的两代人中,有七名男性的面部、手、脚和生殖器畸形和身材矮小。第二年,Scott也描述了一个类似的病例,因此,获得了这个名字:Aarskog-Scott综合征。
这是一种由FGD1基因突变引起的X连锁疾病,因此男性表现出所有的临床表现,而女性携带者只表现出轻微的疾病表现,尤其是面部和手部。
一、一般概述
Aarskog 综合征是一种罕见的遗传病,其特征是身材矮小和多种面部、四肢和生殖器异常。 此外,有时可能会出现某些类型的认知障碍。 到目前为止,X 染色体上的 FGD1 基因是唯一已知与 Aarskog 综合征相关的基因。
二、症状与体征
Aarskog 综合征主要影响男性。受影响的男孩表现出一组特征性的面部、骨骼和生殖器异常。临床症状可能因人而异(临床异质性),即使在家庭内部也是如此。患有 Aarskog 综合征的男性通常有圆脸和宽前额。其他特征性面部特征包括眼睛间距宽(眼距过远)、眼睑下垂(下垂)、眼睑皱襞向下倾斜(睑裂)、鼻孔向前张开的小鼻子(前倾鼻孔)、不发达的上颌骨(上颌骨)发育不全)和寡妇的高峰。受影响的人也可能在上唇(人中)有一个异常长的凹槽和一个宽阔的鼻梁。

图 身材矮小。

图:大鼻子,下垂的上眼睑,肥大和外翻的下唇。
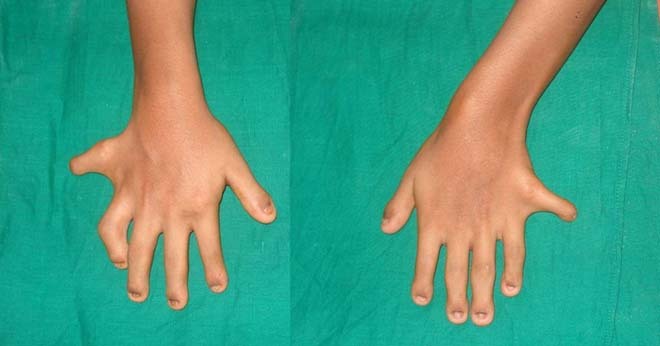
图 双手额外的手指,小指甲。
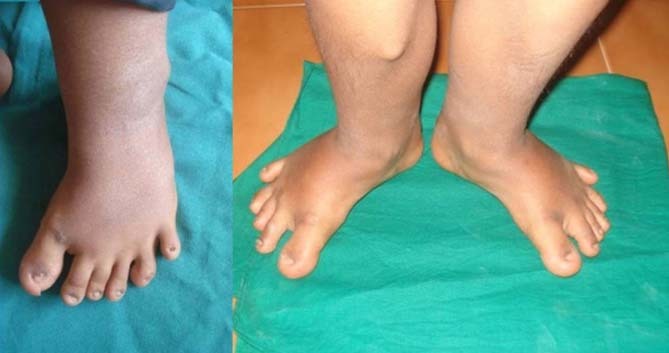
图 弯曲的腿和额外的脚趾。
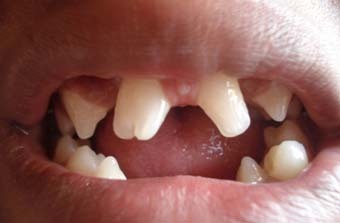
图 中切牙形态改变,中线间隙和下颌中切牙和侧切牙缺失。 增生的唇系带和锥形犬齿
这些孩子也可能有各种影响耳朵和牙齿的异常情况。耳朵异常包括低位耳朵和增厚的“肉质”耳垂。牙齿异常包括出生时牙齿缺失、牙齿萌出延迟和牙齿坚硬外壳发育不全(牙釉质发育不全)。
Aarskog 综合征基本上是一种骨骼发育不良,受影响的男性会出现骨骼系统的特征性畸形,包括不成比例的身材矮小;宽而短的手和脚;短而粗的手指(短指),第五指永久固定在弯曲位置(斜指);手指关节异常伸展;和宽大的扁平足,有球根状的脚趾。此外,受影响的个体可能有凹陷的胸部(漏斗胸)、部分大肠通过腹腔肌肉内层的异常开口突出(腹股沟疝)和突出的肚脐(脐)。患有 Aarskog 综合征的个体可能有脊柱异常,例如脊柱骨骼的不完全闭合(脊柱裂),脊柱上骨(颈椎)融合,以及“钉状”投影发育不全。第二颈椎(齿状突发育不全)。
有助于诊断患有 Aarskog 综合征的男性的体征是生殖器异常,包括围绕阴茎底部延伸的特征性异常皮肤皱襞(“披肩”阴囊)和/或一个或两个睾丸未能下降进入阴囊(隐睾)。此外,尿道口(尿道)可能位于阴茎下侧(尿道下裂),阴囊可能出现裂开或分裂(阴囊分叉)。
在一些受影响的男孩中已经描述了智力障碍,但这并不是该疾病的一致特征。受影响的个体可能会出现一系列轻度学习困难和/或行为障碍:受影响的儿童可能在婴儿期表现出发育迟缓、多动、注意力缺陷、冲动和反对。由于这种可能的特征谱,这种情况也被称为 ADHD 综合征 (MRXS16)。还描述了未能以预期的速度增加体重和生长(未能茁壮成长)以及慢性呼吸道感染的发展。
其他一系列体征和/或症状的发生频率可能较低,包括先天性心脏缺陷;脊柱侧向弯曲异常(脊柱侧弯);额外的肋骨对;上颚闭合不全(腭裂)和/或上唇的垂直凹槽(唇裂);轻微的手指蹼;和有或没有织带的短脖子。可能存在其他眼部异常,包括斜视(斜视)、远视(远视)和某些眼部肌肉麻痹(眼肌麻痹)。据报道,一些患者有超重的倾向。
三、病因
尽管 Aarskog 综合征是一种临床和遗传异质性疾病,但该疾病的最佳特征形式是作为 X 连锁性状遗传,并由 FGD1 基因的变化(突变)引起。 Aarskog 综合征主要影响男性。然而,携带单个 FGD1 基因突变拷贝的女性(杂合子)可能会表现出与该疾病相关的一些症状。在大约 22% 的受累男性中发现了 FGD1 基因突变;因此,其他尚未鉴定的基因也可能与这种情况有关。
四、流行病学
全世界已发表约 60 份经鉴定 FGD1 基因突变证实的 Aarskog 综合征报告。 然而,一些轻度受影响的儿童可能无法识别,因此很难确定一般人群中这种情况的真实频率。 Aarskog 综合征的估计人群患病率等于或略低于 1/25,000。
五、鉴别诊断
以下疾病的症状可能与 Aarskog 综合征的症状相似。比较可能有助于鉴别诊断:
Noonan综合征是一种较为常见的遗传性疾病,以身材矮小、五官畸形和先天性心脏病为特征。这种疾病的特点是症状和身体特征范围广泛,范围和严重程度差异很大。在许多受影响的个体中,相关的异常包括独特的面部外观;宽颈或蹼颈;后发际线低;典型的胸部畸形和身材矮小。头部和面部(颅面)区域的特征性异常可能包括眼睛分布广泛(眼距过远);可能覆盖眼睛内角的皮肤褶皱(内眦褶皱);上眼睑下垂(上睑下垂);小颚(小颌);鼻根凹陷;鼻子短,基部宽;和低位,向后旋转的耳朵(耳廓)。通常还存在明显的骨骼畸形,例如胸骨异常(胸骨)、脊柱弯曲(脊柱后凸和/或脊柱侧凸)和肘部向外偏移(肘外翻)。许多患有 Noonan 综合征的婴儿也有心脏(心脏)缺陷,例如从心脏右下腔到肺部的正常血流受阻(肺动脉瓣狭窄)。其他异常可能包括某些血液和淋巴管的畸形、血液凝固和血小板缺乏、学习困难或轻度智力障碍、受影响的男性在出生后第一年睾丸未能进入阴囊(隐睾),和/或其他症状和发现。 Noonan 综合征可能是由许多基因的突变引起的,包括 PTPN11、KRAS、SOS1、RAF1、NRAS、RIT1 和 SOS2。见:Noonan综合征:症状体征、病因、流行病学、诊断和治疗
Robinow 综合征是一种罕见的遗传性疾病,可以以显性或隐性模式遗传,其特征是由于出生后生长迟缓(出生后生长迟缓)导致的轻度至中度身材矮小;头部和面部(颅面)区域的明显异常;额外的骨骼畸形;和/或生殖器异常。 Robinow 综合征婴儿的面部特征类似于八周大的胎儿。在医学文献中,这种情况通常被称为“胎儿脸”。颅面特征可能包括异常大的头部(大头畸形)和凸出的前额(前额凸起);眼睛间距过大(眼距过远)异常突出;小而上翘的鼻子,鼻孔向前张开(前倾);和/或凹陷(凹陷)的鼻梁。骨骼畸形可能包括异常短的前臂骨(桡骨和尺骨)(前臂短肢)、异常短的手指和脚趾、第五指永久固定在弯曲位置(斜指)、异常小的手和宽拇指、畸形肋骨、脊柱侧向弯曲异常(脊柱侧弯)和/或脊柱中部(胸椎)一侧骨骼(半椎骨)发育不全。与 Robinow 综合征相关的生殖器异常可能包括异常小的阴茎(小阴茎)和受影响男性的睾丸未能下降到阴囊(隐睾)以及阴蒂发育不全(发育不全)和两侧的外部拉长皮肤皱襞受影响女性的阴道开口(大阴唇)。症状的范围和严重程度因人而异。 Robinow 综合征可能是由不同基因的突变引起的,例如 WNT5A、ROR2、DVL3 和 DVL1
Noonan 多发性痣综合征 (NSML) 是一种罕见的遗传性疾病,其特征是皮肤、心脏、内耳、头部和面部(颅面)区域和/或生殖器的结构和功能异常。在患有这种疾病的个体中,症状和身体特征的范围和严重程度可能因人而异。 LEOPARD 是与疾病相关的特征性异常的首字母缩写词:L 代表 (L)entigines(皮肤上出现多个黑色或深褐色斑点); (E)心电图传导缺陷(电活动异常和心脏适当收缩的协调); (0) 眼距过远(眼距宽); (P) 肺动脉狭窄(心脏右心室血液正常流出受阻); (A) 生殖器异常; (R) 生长迟缓导致身材矮小; (D) 由于内耳功能障碍(感觉神经性耳聋)导致的耳聋或听力损失。一些受影响的个体还可能表现出轻度智力障碍、言语困难和/或在某些情况下,还有其他身体异常。 NSML 是一种常染色体显性遗传病。 NSML 和 Noonan 综合征都是由 PTPN11 和 RAF1 基因突变引起的。见:Noonan综合征:症状体征、病因、流行病学、诊断和治疗
六、诊断
Aarskog 综合征的诊断可能基于全面的临床评估、详细的患者和家族史以及特征性发现的识别。 FGD1基因突变的分子遗传学检测可用于确诊。 如果未鉴定出 FGD1 基因突变,可能会建议对与类似情况相关的基因进行分子遗传学检测,例如与 Robinow 综合征相关的 ROR2 和 WNT5A 基因。
从单基因的经典测序到下一代测序 (NGS) 方案的过渡,建议至少使用除 FGD1 外,还包括导致重叠条件的基因的面板,例如 ROR2、WNT5A、PIK3R1、SRCAP、 KMT2D、KDM6A、SHOX、CUL7。
七、治疗
Aarskog 综合征的治疗针对每个人明显的特定症状。治疗可能需要专家团队的协调努力。儿科医生、外科医生、心脏病专家、牙科专家、言语病理学家、评估和治疗听力问题的专家(听力学家)、眼科专家和其他医疗保健专业人员可能需要系统和全面地计划受影响儿童的治疗。
有时可能需要手术来治疗与 Aarskog 综合征相关的特定先天性或结构性畸形(尿道下裂、腹股沟或脐疝、隐睾、异常严重的颅面特征)。 Aarskog 综合征患者应接受完整的眼科和牙科评估。据报道,生长激素治疗可以提高一些儿童的身高,但需要确认以确定适当的管理和对反应的期望。对于可能的神经发育症状,可能需要进行神经精神评估和输入。其他治疗是对症治疗和支持治疗。
建议对受影响的个人及其家庭进行遗传咨询,以明确其家庭中该病的遗传和临床特征、遗传和复发风险。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Ahmed A, Mufeed A, Ramachamparambathu AK, Hasoon U. Identifying Aarskog Syndrome. J Clin Diagn Res. 2016;10(12):ZD09‐ZD11. doi:10.7860/JCDR/2016/22180.8982
Orrico A, Galli L, Clayton-Smith J and Fryns J-P: Clinical utility gene card for: Aarskog–Scott Syndrome (faciogenital dysplasia) – update 2015. Eur J Hum Genet. 2015 Apr; 23(4).
Al-Semari A, Wakil SM, Al-Muhaizea MA et al: Novel FGD1 variant underlying Aarskog-Scott Syndrome with myopathy and distal arthropathy. Clin Dysmorphol. 2013;22:13–17.
Verhoeven WM, Egger JI, Hoogeboom AJ: X-linked Aarskog syndrome: report on a novel FGD1 gene variant. Executive dysfunction as part of the behavioural phenotype. Genet Couns 2012;23:157–167.
Pilozzi-Edmonds L1, Maher TA, Basran RK et al: Fraternal twins with Aarskog-Scott Syndrome due to maternal germline mosaicism. Am J Med Genet A 2011; 55A:1987–1990.
Orrico A, Galli L, Faivre L et al: Aarskog-Scott Syndrome: clinical update and report of nine novel variants of the FGD1 gene. Am J Med Genet A 2010;152A:313–318.
Bottani A, Orrico A, Galli L et al: Unilateral focal polymicrogyria in a patient with classical Aarskog-Scott Syndrome due to a novel missense variant in an evolutionary conserved RhoGEF domain of the faciogenital dysplasia gene FGD1. Am J Med Genet A 2007;143A:2334–2338.
Orrico A, Galli L, Obregón MG, de Castro Perez MF, Falciani M, Sorrentino V: Unusually severe expression of craniofacial features in Aarskog-Scott Síndrome due to a novel truncating variant of the FDG1 gene. Am J Med Genet A 2007;143:58–63.
Kaname T, Yanagi K, Okamoto N, Naritomi K: Neurobehavioral disorders in patients with Aarskog-Scott Syndrome affected by novel FGD1 variants. Am J Med Genet A 2006;140:1331–1332.
Satoh M, Yokoya S: Anabolic steroid and gonadotropin releasing hormone analog combined treatment increased pubertal height gain and adult height in two children who entered puberty with short stature. J Pediatr Endocrinol Metab 2006;19:1125–1131.
Shalev SA, Chervinski E, Weiner E, Mazor G, Friez MJ, Schwartz CE: Clinical variation of Aarskog syndrome in a large family with 2189delA in the FGD1 gene. Am J Med Genet A 2006;140:162–165.
Orrico A, Galli L, Buoni S et al: Attention-deficit/hyperactivity disorder (ADHD) and variable clinical expression of Aarskog-Scott Syndrome due to a novel FGD1 gene variant (R408Q). Am J Med Genet A 2005;135:99–102.
Orrico A, Galli L, Cavaliere ML et al: Phenotypic and molecular characterisation of the Aarskog syndrome: a survey of the clinical variability in light of FDG1 variant analysis in 46 patients. Eur J Hum Genet. 2004;12:16–23.
Lebel RR, May M, Pouls S, Lubs HA, Stevenson RE, Schwartz CE: Non-syndromic X-linked mental retardation associated with a missense variant (P312L) in the FGD1 gene. Clin Genet. 2002;61:139–145.
Schwartz CE, Gillessen-Kaesbach G, May M et al: Two novel variants confirm FDG1 is responsible for the Aarskog syndrome. Eur J Hum Genet 2000;8:869–874.
Pasteris NG, Buckler J, Cadle AB, Gorski JL: Genomic organization of the faciogenital dysplasia (FGD1; Aarskog syndrome) gene. Genomics 1997;43:390–394.
Fryns JP: Aarskog syndrome: the changing phenotype with age. Am J Med Genet. 1992;43:420–427.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#流行病#
43
#综合征#
67