戈谢病:症状与体征、病因、流行病学、诊断与治疗
2023-12-23 MedSci原创 MedSci原创 发表于上海

戈谢病(Gaucher disease,GD) 是较常见的溶酶体贮积病,为常染色体隐性遗传病。
戈谢病(Gaucher disease,GD) 是较常见的溶酶体贮积病,为常染色体隐性遗传病。该病由于葡萄糖脑苷脂酶基因突变导致机体葡萄糖脑苷脂酶(又称酸性 β- 葡萄糖苷酶) 活性缺乏,造成其底物葡萄糖脑苷脂在肝、脾、骨骼、肺,甚至脑的巨噬细胞溶酶体中贮积,形成典型的贮积细胞即“戈谢细胞”,导致受累组织器官出现病变,临床表现多脏器受累并呈进行性加重。又称葡萄糖脑苷脂病、高雪氏病、家族性脾性贫血、脑甙病、脑苷脂网状内皮细胞病等。
戈谢病是一种罕见的遗传性代谢疾病,其中葡萄糖脑苷脂酶的缺乏会导致有害量的某些脂肪(脂质),特别是糖脂葡萄糖脑苷脂,在全身尤其是骨髓、脾脏和肝脏中积累。与戈谢病相关的症状和身体检查结果因患者而异。有些人很少或没有症状(无症状);其他人可能有严重的并发症。
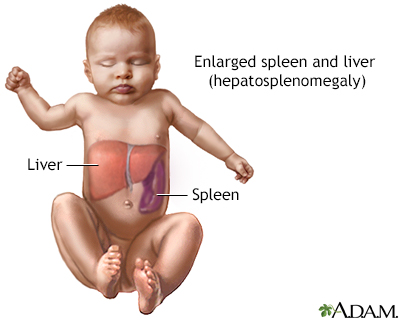
戈谢病的常见表现包括肝脏和/或脾脏异常肿大(肝脾肿大)、循环红细胞水平低(贫血)、血小板水平低(血小板减少症)和骨骼异常。血小板是促进凝血的血细胞,血小板减少症患者可能会出现出血问题。已经确定了三种不同形式的戈谢病,并通过神经系统并发症的不存在或存在和程度来区分。所有三种形式的戈谢病都以常染色体隐性遗传方式遗传。
一、一般概述
戈谢病被归类为溶酶体贮积症(LSD)。 溶酶体是细胞中主要的消化单位。 溶酶体中的酶分解或“消化”营养物质,包括某些复杂的碳水化合物和脂肪。 在戈谢病中,由于缺乏葡萄糖脑苷脂酶,某些含有脂肪的糖(葡萄糖)(称为糖脂)会异常积聚在体内。 这种脂质的积累或“储存”会导致与溶酶体贮积病相关的各种症状或身体检查结果。 戈谢病是第二种最常见的溶酶体贮积症。
二、症状与体征
研究人员已经确定了三种不同形式的戈谢病,由神经系统并发症的缺失(1 型)或存在和程度(2 型或 3 型)分开。其他形式的戈谢病包括围产期致死形式和心血管形式。戈谢病患者的具体症状因人而异。有些人表现出很少或没有症状(无症状);其他人经历慢性,有时是严重的并发症。
戈谢病 1 型也称为非神经元病,成人型,因为它不涉及中枢神经系统(大脑和脊髓)。 1 型戈谢病是该病最常见的形式。大多数患有 1 型戈谢病的人由于称为血小板的凝血细胞水平低(血小板减少症)容易瘀伤,由于循环红细胞水平低导致慢性疲劳(贫血),以及肝脏和/或脾脏异常肿大。肝脾肿大)。受影响的个体还可能经历身体各种骨骼的血液供应不足(梗塞),导致钝痛或剧烈的骨骼疼痛(骨骼危机)、退化(缺血性坏死)和受影响骨骼的畸形,以及骨骼变薄和变弱(骨质疏松症) .这种骨骼异常导致骨折的易感性增加。在极少数情况下,受影响的个体也可能会经历肺部和/或肾脏的受累。
2型戈谢病,也称为急性神经元性戈谢病,发生在新生儿和婴儿身上,其特征是由于大脑中葡萄糖脑苷脂的异常积聚导致的神经系统并发症。脾脏肿大(脾肿大)通常是首发症状,并且可能在六个月大之前变得明显。肝脏肿大(肝肿大)并不总是很明显。受影响的婴儿可能会失去先前获得的运动技能,并表现出低肌张力(肌张力减退)、不自主肌肉痉挛(痉挛),导致手臂和腿缓慢、僵硬的运动,以及斜视(斜视)。此外,受影响的婴儿可能会出现吞咽困难(吞咽困难),这可能导致喂养困难;颈部异常定位或弯曲(后屈);由于喉部肌肉收缩(喉痉挛),体重增加和生长速度未能达到预期(无法茁壮成长)和高音调的呼吸(喘鸣)。也可能出现贫血和血小板减少症。 2 型戈谢病通常会发展为危及生命的并发症,例如呼吸窘迫或食物进入呼吸道(吸入性肺炎)。严重受影响的新生儿可能会出现皮肤异常(火棉胶皮肤或鱼鳞状变化)和全身肿胀(水肿),并在出生后的最初几周内死亡。其他患有 2 型戈谢病的儿童的寿命大大缩短,死亡通常发生在 1 到 3 岁之间。
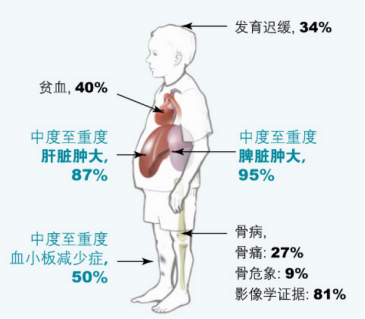
3 型戈谢病,也称为慢性神经元性戈谢病,幼年型,发生在生命的头十年。除了上面讨论的血液和骨骼异常外,受影响的个体还会出现神经系统并发症,其发展和进展比戈谢病 2 型慢。相关的神经系统并发症包括精神恶化;无法协调自主运动(共济失调);手臂、腿或全身的短暂、类似休克的肌肉痉挛(肌阵挛发作)。一些患有 3 型戈谢病的人可能难以左右移动他们的眼睛(水平凝视麻痹)。患有 3 型戈谢病的患者也可能出现垂直凝视麻痹,该麻痹通常发生在水平凝视麻痹之后。很大一部分患者还会发展为肺(肺)病(间质性肺病)。 3 型戈谢病患者的表现和临床过程可能存在很大差异。一些受影响的患者可能活到十几岁和 20 岁出头,而其他人的寿命更长(30 岁和 40 岁)。随着困难的增加,受影响的个体可能需要帮助来完成日常生活任务(例如,吃饭、洗澡和行走)。

围产期致死形式或胎儿/新生儿戈谢病发生在不到 5% 的患者中。这种类型非常严重,并且与 3 个月大之前甚至在子宫内的死亡有关。胎儿/新生儿可能出现广泛的皮肤肿胀(水肿或全身水肿),导致心脏、皮肤或肺部积液(胎儿水肿)。其他症状包括颅内出血(颅内出血)、皮肤脱屑(非大疱性鱼鳞状红肿),外观呈红色,以及关节在固定弯曲位置收缩(先天性多发性关节弯曲)。
心血管形式的特征在于 CNS 受累,例如难以在所需方向上开始眼球运动(动眼神经失用症)。其他症状包括二尖瓣和主动脉瓣钙化、角膜混浊和轻度脾肿大。心脏上的钙沉积物会减少流向这些瓣膜的血流量,并会增加血压。也可能存在核上性眼肌麻痹,这会导致平衡、行走和思维问题。与心脏相关的并发症和相关的神经系统问题会导致寿命缩短,尽管在某些患者中这可能会延续到青年期。
三、病因
戈谢病是由 GBA 基因的变化(突变)引起的。
所有三种形式的戈谢病都以常染色体隐性遗传方式遗传。 包括经典遗传疾病在内的人类特征是两个基因相互作用的产物,一个来自父亲,一个来自母亲。
四、流行病学
所有形式的戈谢病都会影响同等数量的男性和女性。戈谢病 1 型是最常见的类型,占高加索人病例的 90% 以上。患有 1 型戈谢病的人通常在青春期表现出症状,但发病年龄从儿童期到成年期不等。戈谢病 2 型的发病年龄是婴儿早期。 3 型戈谢病的发病年龄各不相同,但该病通常始于儿童期或青春期。戈谢病的神经性形式的频率,即此类病例的比例,在非高加索人中较高。
GD 患病率全球各地区不尽相同。一项系统分析统计全球每 10 万人中发病人数为0.7~1.75,是全球范围内最为常见的溶酶体贮积疾病之一。有德系犹太人血统的(系指中欧及东欧犹太人) 人群发病率最高,每 450 个德系犹太婴儿中就有 1 个患病。一项国内的人口统计研究发现,中国东部人口中 GD 是排名第 4 的溶酶体贮积疾病。国内较为准确的GD 发病率研究来自于上海一项以干血斑法筛查新生儿葡萄糖脑苷酯酶活性,发现 GD 的发病率为 1 :80 844。瑞典北博滕省人口的估计患病率为 50,000 分之一。但是,在瑞典的北博滕地区,有一种 3 型戈谢病亚型发生率更高(诺尔博特尼亚戈谢病)。中国内地尚没有建成全国性的 GD 登记中心,全面的流行病学调查有待完善。
五、鉴别诊断
以下疾病的症状可能与戈谢病的症状相似。比较可能有助于鉴别诊断:
Niemann-Pick 病 (NPD) 是一组罕见的脂肪代谢遗传性疾病。已确定至少五种类型的尼曼-皮克病(NPD 类型 A、B、C、D 和 E)。 A 型和 B 型症状是由于缺乏酸性鞘磷脂酶 (ASM) 而导致的,该酶是分解鞘磷脂所必需的,鞘磷脂是一种存在于所有组织中的脂肪物质,尤其是在大脑和神经系统中。这种缺乏会导致身体的许多器官(如肝脏、脾脏和大脑)中过量的鞘磷脂异常积累。 C型症状的出现是因为无法调动胆固醇和其他物质,导致身体各个器官中这些物质过量。所有类型尼曼-皮克病的常见症状包括皮肤、眼睛和/或黏膜黄染(黄疸)、运动技能的进行性丧失、喂养困难、学习障碍以及肝脏和/或脾脏异常肿大。肝脾肿大)。不同类型的尼曼-匹克病作为常染色体隐性遗传。
庞贝病是一种糖原贮积病。这种遗传性代谢紊乱是由先天性缺乏酶 α-1,4 葡糖苷酶(溶酶体葡糖苷酶;酸性麦芽糖酶)引起的,这种酶是分解糖原所必需的,糖原是身体的能量来源。这种酶缺乏导致过量的糖原在溶酶体中积累,溶酶体是细胞内分解细胞内废物的结构。庞贝病的症状和身体表现是由细胞中糖原的异常积累引起的。已经确定了三种不同形式的庞贝病。婴儿型的特征是严重的肌肉无力和肌张力异常减弱(肌张力减退),但没有肌肉萎缩,通常在出生后的头几个月内表现出来。其他异常可能包括心脏(心脏肿大)、肝脏(肝肿大)和/或舌头(巨舌)肿大。进行性心力衰竭通常会在 12 至 18 个月大时引起危及生命的并发症。童年形式通常在婴儿后期或儿童早期开始。受累个体的器官受累程度可能不同;然而,骨骼肌无力通常伴随着最小的心脏受累。在庞贝病的成人形式中,症状包括肌肉无力,例如在其他慢性肌肉疾病中发现的。症状的发作通常发生在第二到第四个十年。这种形式的疾病进展缓慢,没有心脏受累,但可能与严重的肺部并发症(呼吸衰竭)有关。
Hurler 综合征 (MPS I) 是一组称为粘多糖症 (MPS 疾病) 的疾病之一,这是一种罕见的遗传性疾病,由十种特定溶酶体酶中的一种缺乏引起,导致无法分解复杂的碳水化合物 (粘多糖)变成更简单的分子。这些大的、未降解的粘多糖(也称为糖胺聚糖)在身体细胞中的积累会导致几种身体症状和异常。存在三种严重程度不同的 Hurler 综合征。患有 Hurler 综合征的婴儿通常在出生时表现正常,但可能有腹股沟和脐疝、角膜混浊、肝脾肿大、舌头大、骨骼异常、生长不良和关节僵硬。 Hurler 综合征是由缺乏 α-L-艾杜糖醛酸酶引起的。
Tay-Sachs 病是一种罕见的神经退行性疾病,其中一种酶(己糖胺酶 A)的缺乏导致大脑中某些称为神经节苷脂的脂肪(脂质)过度积累。这种神经节苷脂的异常积累导致中枢神经系统中细胞的进行性破坏。与 Tay-Sachs 病相关的症状可能包括对突发噪音的过度惊吓反应、精神萎靡、丧失先前获得的技能(即精神运动退化)和肌肉张力严重减弱(肌张力减退)。随着疾病的进展,受影响的婴儿和儿童可能会在眼睛中层(特别是视网膜神经节细胞)出现樱桃红色斑点,逐渐丧失视力和耳聋,增加肌肉僵硬和运动受限(痉挛),最终瘫痪,大脑中不受控制的电干扰(癫痫发作)和恶化
六、诊断
对于不明原因的贫血和容易瘀伤的个体,尤其是脾脏和肝脏肿大和骨折的个体,应考虑诊断为戈谢病。戈谢病的诊断可以通过彻底的临床评估和各种专业测试来确认,特别是测量白细胞(白细胞)或皮肤细胞(成纤维细胞)中酸性 β-葡萄糖苷酶活性的测试(即酶测定)和遗传(DNA)分析因果基因缺陷(突变)。
1. 葡萄糖脑苷脂酶活性检测 葡萄糖脑苷脂酶活性检测是 GD 诊断的金标准。当其外周血白细胞或皮肤成纤维细胞中葡萄糖脑苷脂酶活性降低至正常值的 30%以下时,即可确诊 GD。值得注意的是,少数患者虽然具有 GD 临床表现,但其葡萄糖脑苷脂酶活性低于正常值低限但又高于正常低限 30%时,需参考该患者血中生物学标志物结果(壳三糖酶活性等),进一步做基因突变检测,从而实现确诊。
2. 骨髓形态学检查 大多数 GD 患者骨髓形态学检查能发现特征性细胞即戈谢细胞, 该细胞体积大,细胞核小,部分胞质可见空泡。但该检查存在假阴性及假阳性的情况。当骨髓中查见戈谢细胞时,应高度怀疑 GD,但并不能确诊 GD,需在鉴别区分其他疾病的同时,进一步做葡萄糖脑苷脂酶活性测定。
3. 基因检测 目前已发现的葡萄糖脑苷脂酶基因突变类型有 400 多种,相似的表型可有多种不同基因型,而相同基因型的患者临床表现、病程及治疗效果也不同。葡萄糖脑苷脂酶基因的突变类型具有种族差异,并与临床表型相关。到目前为止,已发现中国人GD 基因突变类型约 40 种,以 L444P 为最常见的突变类型,可出现在有神经系统症状及无神经系统症状的 GD 各型患者中,其次为 F213I、N188S、V375L 和 M416V 突变类型。基因诊断并不能代替酶活性测定的生化诊断,但可作为诊断的补充依据并明确对杂合子的诊断。少数突变与患者的临床分型具有相关性,对判断疾病程度和预后具有指导作用。
4. 如果家族中存在已知的 GBA 基因突变,则可以进行戈谢病的产前诊断。检测可通过羊膜穿刺术或绒毛膜绒毛取样 (CVS) 进行,但不常见,除非有 2 型戈谢病家族史。在羊膜穿刺术期间,取出并分析胎儿周围的液体样本(羊水),而CVS 涉及从胎盘的一部分中去除组织样本。产前诊断可以确定戈谢病的诊断,但不能确定疾病的类型
七、治疗
主要目标是通过让患者进行正常的日常活动来提高他们的生活质量,例如在工作时不会感到过度疲劳或正常行走而不会感到关节疼痛。其他目标包括预防并发症的严重程度,例如骨密度降低至变薄、骨骼脆弱(骨质疏松症)以及肺功能降低导致容易骨折或呼吸急促。使孩子的生长正常化以使其达到正常身高也可以成为治疗后几年内的目标,并实现正常的青春期开始。
根据戈谢病的类型,对每位患者进行个体化治疗。 1 型戈谢病被认为是可治疗且轻微的,因为它不涉及神经系统症状,因为大脑不受影响。由于婴儿时期的快速且不可逆转的脑损伤,目前认为 2 型不可治疗。 3 型仍涉及神经损伤,但这些症状的进展比 2 型慢。目前 FDA 批准的药物治疗选择包括酶替代疗法 (ERT) 和底物减少疗法 (SRT)。
酶替代疗法 (ERT) 已被证明对 1 型戈谢病患者有效。在 ERT 的研究中,贫血和低血小板计数得到改善,肝脏和脾脏的肿大已大大减少,骨骼表现有所改善。这些全身性表现在接受 ERT 的 2 型和 3 型戈谢病患者中也有所改善。然而,ERT 并未有效减少或逆转与 2 型和 3 型戈谢病相关的某些神经系统症状。
ERT 每 2 周通过静脉 (IV) 输注在输液中心、国家戈谢病治疗中心或在家中通过自我给药、家人/朋友或家庭护理护士的协助进行。目前 FDA 批准的三种 ERT 药物包括伊米苷酶 (Cerezyme)、维拉苷酶 alfa (VPRIV) 和塔利苷酶 (Elelyso)。
1991 年,美国食品药品监督管理局 (FDA) 批准了用于治疗 1 型戈谢病的孤儿药阿糖苷酶注射液 (Ceredase),这是第一个被证明对治疗有效的 ERT戈谢病 1 型。
这种药物的合成形式伊米苷酶 (Cerezyme) 于 1994 年获得批准。重组 DNA 技术或基因工程用于生产 Cerezyme。这是克服源自人类胎盘的 Ceredase 可用性限制的重要一步。因此,Ceredase 已退出市场,原因是生产的类似药物没有来自人类细胞的生物利用度问题和疾病转移。由 Genzyme 制造的 Cerezyme 替代了戈谢病患者缺乏的人类溶酶体酶葡萄糖脑苷脂酶。
另一种经 FDA 批准的称为 Velaglucerase alfa(商品名 VPRIV)的葡萄糖脑苷脂酶制剂可从 Shire 获得。
辉瑞公司的 Elelyso(也称为 Uplyso 或 taliglucerase alfa)在 Protalix BioTherapeutics Inc. 的许可下于 2012 年被 FDA 批准用于治疗 1 型戈谢病。Elelyso 是一种注射的长期酶替代疗法,应由医疗保健专业人员每隔一周进行一次。它使用基因工程胡萝卜细胞来提供替代葡萄糖脑苷脂酶。
底物减少疗法也可用于特定患者群体。它们通过抑制葡萄糖神经酰胺合酶来阻断葡萄糖脑苷脂(脂肪物质)的产生,与 ERT 的工作方式不同。这些是片剂/胶囊剂,每天服用。 SRT 不适用于儿童和青少年、孕妇或哺乳期妇女、老年患者以及患有严重肾脏或肝脏疾病的人。目前 FDA 批准的两种药物包括 eliglustat (Cerdelga) 和 imiglustat (Zavesca)。
2014年,Genzyme公司生产的Cerdelga(eliglustat)被FDA批准用于1型戈谢病成年患者的长期治疗。
2003 年,美国食品和药物管理局批准了口服疗法 Zavesca,用于治疗不能选择酶替代疗法的轻度至中度戈谢病 1 型成年患者(由于过敏、超敏反应等) )。
基因治疗也在研发中,基因疗法PR001治疗神经性戈谢病,目前在研发中。
建议对受影响的个人及其家人进行遗传咨询。其他治疗是对症治疗和支持治疗。
美国国家人类基因组研究所医学遗传学分部目前正在研究戈谢病和帕金森病之间可能存在的联系或关联。研究表明,受影响的个体(具有两个引起疾病的 GBA 基因突变)和携带者(具有单个 GBA 基因突变)都具有增加的帕金森病风险。
患有戈谢病的人患多发性骨髓瘤的风险增加,成人应仔细监测。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
戈谢病多学科诊疗专家共识(2020), https://www.medsci.cn/guideline/show_article.do?id=6636c1c00201e9d8
中华医学会儿科学分会遗传代谢内分泌学组,中华医学会儿科学分会血液学组,中华医学会血液学分会红细胞疾病(贫血)学组 . 中国 GD 诊治专家共识 . 中华儿科杂志,2015,53(4):256-261.
Coles V., Chan G., Palczewski K., Lewis K., Ho J. An Unexpected Link Between Gaucher Disease and Parkinson’s Disease. Illustrated by Cindy Nguyen. Rare Disease Review. March 2018. DOI:10.13140/RG.2.2.27808.07680.
FDA Prescription and Over-the-Counter Drug Product List. 32ND Edition Cumulative Supplement Number 3: March 2012.
Khan, Mohammed, et al. Gaucher’s Disease: Prenatal and Post Natal Diagnostic Dilemma and Biochemical Aid – Case Series and Review of Literature. British Journal of Medicine and Medical Research. 2017;19(5): 1-11. doi:10.9734/bjmmr/2017/29680.
Substrate Reduction Therapy (Oral Medication) for Gaucher Disease.” National Gaucher Foundation, www.gaucherdisease.org/gaucher-diagnosis-treatment/treatment/substrate-reduction/. Accessed September 10, 2018.
Enzyme Replacement Therapy for Gaucher Disease.” National Gaucher Foundation, www.gaucherdisease.org/gaucher-diagnosis-treatment/treatment/enzyme-replacement-therapy/. Accessed September 10, 2018.
Özkaya, Özge. Managing Gaucher Disease: New Set of Goals Established by Experts. Gaucher Disease News. 14 Mar. 2017, gaucherdiseasenews.com/2017/03/14/new-goals-established-management-gaucher-disease/. Accessed September 10, 2018.
National Institute of Neurological Disorders and Stroke: Gaucher Disease Information Page.Last updated 6/22/2018. Available at https://www.ninds.nih.gov/Disorders/All-Disorders/Gaucher-Disease-Information-Page Accessed September 10, 2018.
Gaucher disease-Genetics Home Reference. Reviewed September 2014. Available at: https://ghr.nlm.nih.gov/condition/gaucher-disease Accessed September 10, 2018.
Gaucher disease. Genetic and Rare Diseases Information Center. Last updated: 10/28/2017. https://rarediseases.info.nih.gov/diseases/8233/gaucher-disease. Accessed September 10, 2018.
Pastores GM, Hughes DA. Gaucher Disease. 2000 Jul 27 [Updated 2018 Jun 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1269/ Accessed September 10, 2018.
重磅|思而赞®在华获批可用于Ⅲ型戈谢病治疗, https://news.bioon.com/article/6710799.html
基因疗法PR001治疗神经性戈谢病:IND已生效 https://www.medsci.cn/article/show_article.do?id=f6ec1853363a
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#诊断与治疗#
58
#流行病#
45
平时都没注意的这个疾病,学到了
47