
一、概述 参考文献: [1] Priya S, Kishnani, Stephanie L. Austin, Jose E. Abdenur. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med, 2014,16(11):e1.
二、病因和流行病学
三、临床表现
四、辅助检查
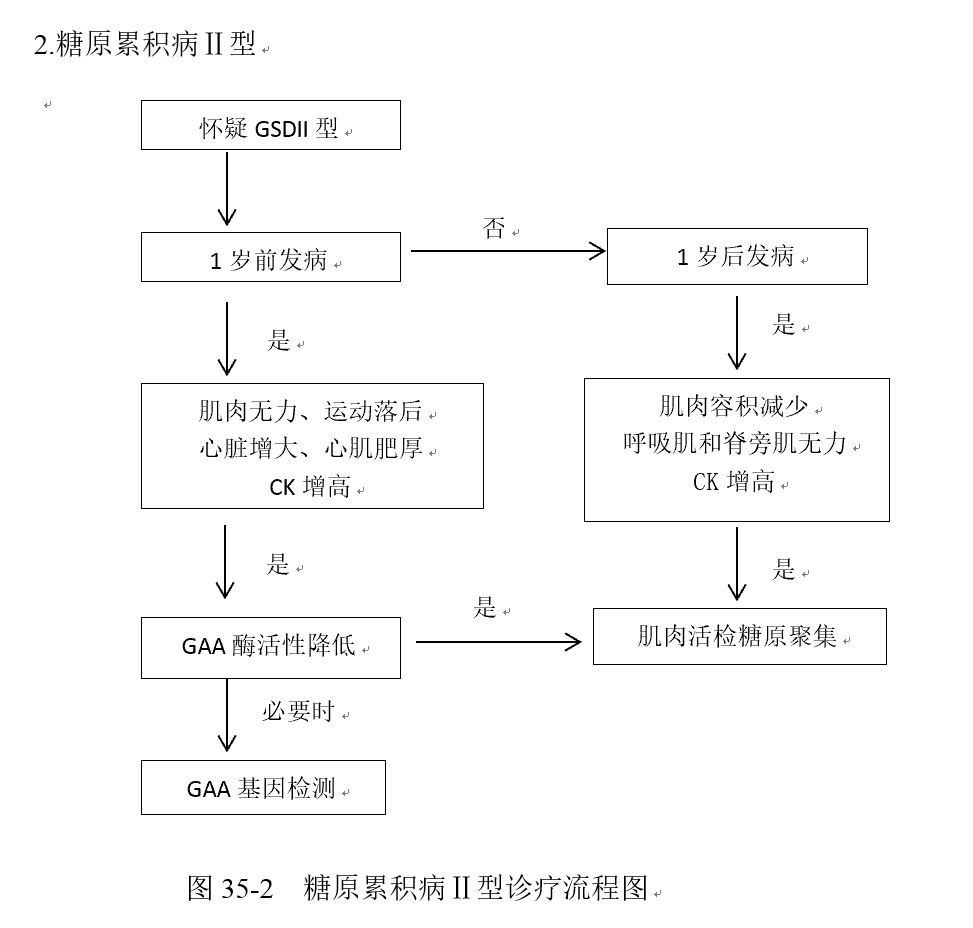
五、诊断与鉴别诊断
六、治疗
[2] http://www.ncbi.nlm.nih.gov/sites/genetests
[3] 邬玲仟,张学.医学遗传学.北京:人民卫生出版社,2016:362-368.
[4] Van der Meijden JC, Gunggor D, et al. Ten years of the international Pompe survey: patient reported outcomes as a reliable tool for studying treated and untreated children and adults with non-classic Pompe disease. J Inherit Metab Dis, 2015, 38(3):495-503.
[5] 中华医学会儿科学分会内分泌遗传代谢学组,中华医学会儿科学分会神经学组,中华医学会神经病学分会肌电图与临床神经生理学组,中华医学会神经病学分会神经肌肉病学组.糖原贮积病型诊断及治疗专家共识.中华医学杂志,2013,93(18):1370-1373.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#糖原累积病#
34